Single-Molecule Electronics @ UoL
Behind the Paper
The stuff I couldn't fit in the manuscript
17/06/2026
Charge Transport through Carbon Atomic Chains
Not too bad I'd say... Only 16 months since the last "behind the paper"! I must admit we've been quite busy - lots of good news in the meanwhile, lots of bad
news (hey, it's academia), a few teary farewells and some warm welcomes. But I'm not here to tell you about these. I'm here because we somehow convinced the editors of
Nature Chemistry and four reviewers that our madness made sense, and I want to tell the story of how we got there.
It's long and full of experiments, magnificent unicorns, people who still have 10 fingers, and the work of two absolutely brilliant students (plus
a corollary of 10+ people that helped out). Put your safety goggles on, close the sash of the fume cupboard and - for good measure - get behind the blast shield.
Today we'll talk about carbon chains!

This story starts with a "care package" coming all the way from Australia. Paul Low (if you're not familiar with him/his work, see below for Saman's story also involving a mysterious Australian package) and his student Jarred sent us some compounds with a quirky note attached: "These seem to work but they're weird. See what you make of them." which translates from academic to plainspeak to "We've tried everything with these, and now it's your turn to be confused". I took a look at the structures and as I realised what they were trying to do, a large smile spread on my face.
 Now quite a key thing happened: James (see his smiling
face here when he was still a strapping young lad and not the grizzled Dr. Morris he has now become) was already measuring some thioanisolyl-capped oligoynes for another
project, so he got the proverbial short straw. He took these Au(I) complexes, measured them, did a bunch of analysis, modelling and control experiments, and after a year or so of work
(which includes a full recode/rebuild of the STMBJ machine, from Leviathan Mk.II to Leviathan Mk.III, masterfully accomplished by James and Tom - more on this later) and multiple rounds of data acquisition we had a
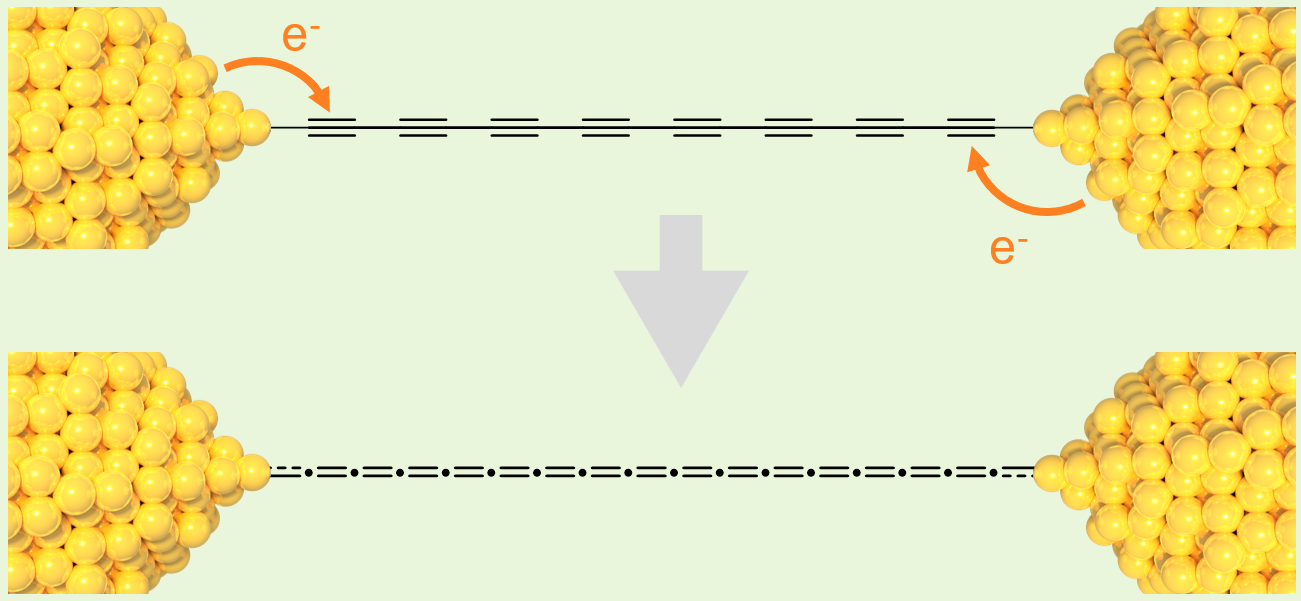
nice little study on our hands. Our theory then was that, as the junction evolves, we are forming coordination polymers by incorporating small Au(0) clusters - extruded from the electrodes - into
the junction. Something like the cartoon below, a phenomenon already observed in 1,4-dicyanobenzene on Au electrodes
and 1,4-diethynylbenzene on Ag electrodes.
Now quite a key thing happened: James (see his smiling
face here when he was still a strapping young lad and not the grizzled Dr. Morris he has now become) was already measuring some thioanisolyl-capped oligoynes for another
project, so he got the proverbial short straw. He took these Au(I) complexes, measured them, did a bunch of analysis, modelling and control experiments, and after a year or so of work
(which includes a full recode/rebuild of the STMBJ machine, from Leviathan Mk.II to Leviathan Mk.III, masterfully accomplished by James and Tom - more on this later) and multiple rounds of data acquisition we had a
nice little study on our hands. Our theory then was that, as the junction evolves, we are forming coordination polymers by incorporating small Au(0) clusters - extruded from the electrodes - into
the junction. Something like the cartoon below, a phenomenon already observed in 1,4-dicyanobenzene on Au electrodes
and 1,4-diethynylbenzene on Ag electrodes.
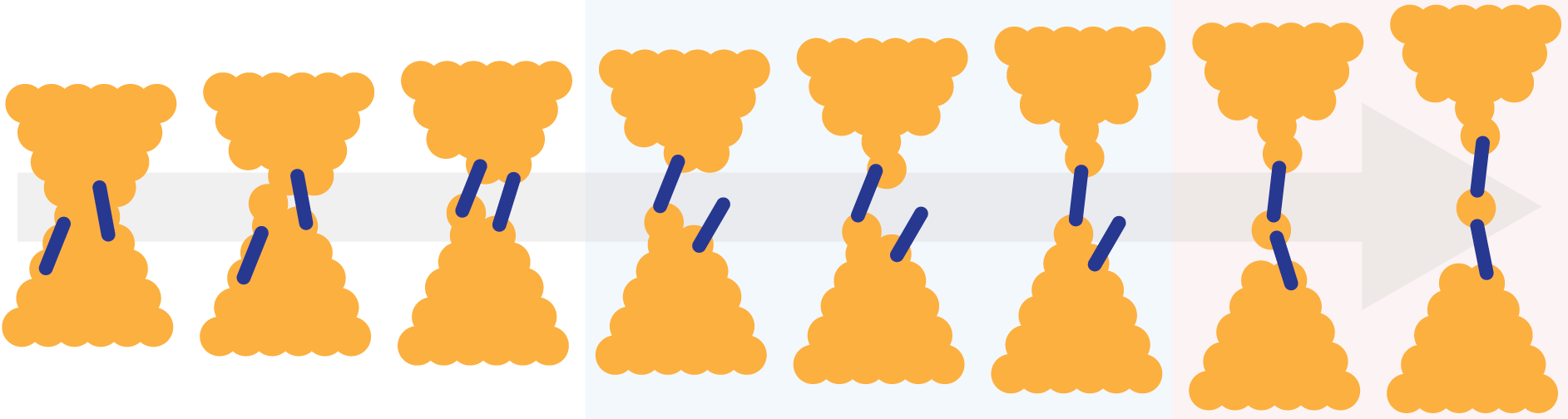
A bit of a "chronological digression" here... I had to scour my emails to reconstruct the timeline of this project, and it looks like the first "care package" arrived in Liverpool in July 2022, we worked on and off with them for a while, and we had sort-of-finalised the paper about this first batch of compounds in June 2024. It was summer, I had very few commitments and I always enjoy an excuse to go back to the lab. I measured the pentayne (C10) and it fitted our model perfectly. C12, however, was a different beast altogether: it showed a quite significant boost in conductance, going even higher than the very short C4. Now that was very interesting... It's time to stop the narration for a bit and discuss the science.
 When you have a pure ...CCCCCCCCC... carbon chain, there are two possible valence bond description. One that has alternating triple and single bonds (polyyne, ...C≡C-C≡C...)
while the other has a continuous double-bond pattern (cumulene, ...C=C=C=C...). An infinite polyyne is still semiconducting, with a definite HOMO-LUMO gap, while the corresponding
cumulene is predicted to be metallic. Making such "infinite" (or at least very long) carbon chains is one of the holy grails of materials chemistry: this is the material
called carbyne (also known as Linear Acetylenic Carbon - LAC) that has been predicted to have amazing electrical, thermal, spectroscopic, and mechanical properties. The full shebang: stronger than graphene, harder than diamond, more
conductive than gold, etc. It's however - as of 2026 - all speculation. Carbyne is an elusive and potentially very dangerous material: only weeks before his death, Harry Kroto said
"I know they have not made carbyne because they're still alive" in reply to a claim by a Chinese team of having made this holy-grail material. And that's because carbon
chains - whether in their polyyne or cumulene structure - become violently reactive as they are made longer: adjacent chains will spontaneously crosslink, with intense release
of energy. It's essentially a high-velocity explosive. This danger however has not prevented many experimental groups from trying to make long carbon chains: from Rik
Tykwinsky, holding the current record for a 24-yne (a string of 48 carbon atoms) stabilised with bulky organic capping groups,
to the encapsulation methods pioneered by Tomoko Suzuki, such a simple string of atoms keeps captivating the interest of chemists and physicists around the world.
When you have a pure ...CCCCCCCCC... carbon chain, there are two possible valence bond description. One that has alternating triple and single bonds (polyyne, ...C≡C-C≡C...)
while the other has a continuous double-bond pattern (cumulene, ...C=C=C=C...). An infinite polyyne is still semiconducting, with a definite HOMO-LUMO gap, while the corresponding
cumulene is predicted to be metallic. Making such "infinite" (or at least very long) carbon chains is one of the holy grails of materials chemistry: this is the material
called carbyne (also known as Linear Acetylenic Carbon - LAC) that has been predicted to have amazing electrical, thermal, spectroscopic, and mechanical properties. The full shebang: stronger than graphene, harder than diamond, more
conductive than gold, etc. It's however - as of 2026 - all speculation. Carbyne is an elusive and potentially very dangerous material: only weeks before his death, Harry Kroto said
"I know they have not made carbyne because they're still alive" in reply to a claim by a Chinese team of having made this holy-grail material. And that's because carbon
chains - whether in their polyyne or cumulene structure - become violently reactive as they are made longer: adjacent chains will spontaneously crosslink, with intense release
of energy. It's essentially a high-velocity explosive. This danger however has not prevented many experimental groups from trying to make long carbon chains: from Rik
Tykwinsky, holding the current record for a 24-yne (a string of 48 carbon atoms) stabilised with bulky organic capping groups,
to the encapsulation methods pioneered by Tomoko Suzuki, such a simple string of atoms keeps captivating the interest of chemists and physicists around the world.
While the chemistry of these chains is the work of mavericks, the physics behind them is quite well studied and - more or less - understood. While short carbon chains could adopt the metallic, cumulene-like structure ..C=C=C=C... once the length reaches a critical value a phenomenon called "Peierls' distortion" kicks in, favouring the polyyne structure ...C≡C-C≡C... and therefore stabilising a semiconducting behaviour. Key here is a parameter called "Bond Length Alternation" (BLA), that describes the extent of polyyne character: a perfect cumulene has all double bonds equal in length, and hence BLA = 0. Non-zero values of BLA are indicative of a degree of polyyne character, up to a limit value of ~0.2 Å found in butadiyne. An infinite carbon chain, due to Peierls' distortion, should have BLA to settle around 0.13 Å - a quite strong alternation of short (triple) and long (single) bonds. The shorter synthetic carbon chains, bearing bulky stabilising groups at the two ends to prevent explosive decomposition, can be made in their polyyne or cumulene form by accurate choice of the capping group and synthetic procedure: the nature of the capping groups determines the structure and, hence, the BLA.
Here's the catch: nobody knows what happens when the capping groups are massive, macroscopic metallic electrodes. I hope you now understand why a smile spread on my face when I saw the structures of the compounds sent by Paul & Jarred. We were looking for an effect of the Au electrodes on the carbon chains, and it looks like we finally found it. While spectroscopically (UV, Raman, etc.) the Au(I) complex of C12 did not behave in any significantly different way than its shorter brothers and sisters, something definitely changed once it was "soldered" (or, more precisely, once it transmetallated) to the two Au electrodes. These data suggested that the Au electrodes were forcing an electronic equalisation of the carbon chain, from the semiconducting ...C≡C-C≡C... to the more conductive, quasi-metallic ...C=C=C=C... valence structure.
 Now that's a big claim, and to defend it we immediately realised we needed three things: more data, more/better modelling/theory,
and an independent proof beyond the STMBJ measurements.
The results on C12 sent James into a flurry of activity: he had two months to rewrite an entire chapter of his thesis, and he also decided that the theoretical tool we had used so far (ANT)
was not good enough: he had to write his own (the Tight-Binding Funtime Engine, something that is evolving from a simple funny tool into a fully-fledged computational suite that is slowly eroding
James' sanity - but this story is for another day). Fair. Just as things started to settle again, Jarred sent over the C16 analogue and - shortly after - the synthetically
trickier C14 compound. Now a small side-note
on Jarred. Neither James nor I had ever seen him at this point in time. Given the 14000 km separating us (my brain is metric, but for those of you unaccustomed
to proper units, it's about 9000 miles, or 31818181 cubits), that's not entirely surprising.
Anyway, in our collective imagination, Jarred took on the status of a mythical creature capable of the most magical synthetic feats, undeterred
by the inherent risk of working with the Csp equivalent of TNT, surfing the majestic waves of Western Australia while the NMR acquires the spectrum of the last product he made. Something
like the picture here: a magnificent unicorn.
Now that's a big claim, and to defend it we immediately realised we needed three things: more data, more/better modelling/theory,
and an independent proof beyond the STMBJ measurements.
The results on C12 sent James into a flurry of activity: he had two months to rewrite an entire chapter of his thesis, and he also decided that the theoretical tool we had used so far (ANT)
was not good enough: he had to write his own (the Tight-Binding Funtime Engine, something that is evolving from a simple funny tool into a fully-fledged computational suite that is slowly eroding
James' sanity - but this story is for another day). Fair. Just as things started to settle again, Jarred sent over the C16 analogue and - shortly after - the synthetically
trickier C14 compound. Now a small side-note
on Jarred. Neither James nor I had ever seen him at this point in time. Given the 14000 km separating us (my brain is metric, but for those of you unaccustomed
to proper units, it's about 9000 miles, or 31818181 cubits), that's not entirely surprising.
Anyway, in our collective imagination, Jarred took on the status of a mythical creature capable of the most magical synthetic feats, undeterred
by the inherent risk of working with the Csp equivalent of TNT, surfing the majestic waves of Western Australia while the NMR acquires the spectrum of the last product he made. Something
like the picture here: a magnificent unicorn.
The magnificent unicorn delivered. It was our duty to render proper justice to Jarred's brave (and monumental) efforts. James was deep in thesis-land, so I measured them and they were PERFECT. C16 specifically was an absolute pleasure to observe delivering long and incredibly conductive junctions. Current did not decay AT ALL with length: all three of them - C12, C14, and C16 - had the same conductance. But that alone wasn't enough to demonstrate cumulene equalisation - other less exciting phenomena could explain the observed behaviour. Now, the main thing that happens when the electronic structure of a carbon chain changes from polyyne to cumulene is that the HOMO-LUMO gap (the difference in energy between the last populated orbital and the first empty one) decreases. A perfect cumulene, in fact, is metallic exactly because its HOMO-LUMO gap vanishes completely: like a metal, there is no energy gap between full and empty states. Our calculations showed the reduction in HOMO-LUMO gap pretty well too, and in molecular electronics there is a surefire way of probing it. We have to run I-V curves - measuring how the transported current evolves with the applied bias. When one of the resonances is close to the Fermi level of the electrodes, the current through the single-molecule junction deviates from the traditional behaviour, and shoots up - sometimes quite dramatically.
BACK TO THE LAB!
 At this point in time, James was finally done with his thesis. He took the helm of our side of the project again, and he made himself very busy - I may even have to use the word "obsessed" - over the next few months...
He collected all I-V curves, he analysed GBs and GBs of data, and he put it all together in truly relentless grind, continuously cross-checking and optimising his Funtime Engine.
Now the work he and Tom performed on the STM paid off handsomely. As I mentioned, they worked on a complete rebuild/recode of our flagship instrument The Leviathan (now in its Mk.III
variant and currently undergoing some further changes towards Mk.IV), with the key objective of integrating a PID feedback loop to ensure a completely automatic/unsupervised data collection.
A "press-start-and-forget" approach. In essence, they were tired of babysitting the STM, so they wrote an algorithm that did it for them. Experiments that used to take a full day or even
multiple days now only take a few hours, which means we can quickly explore a wider range of experimental parameters, and easily do very demanding experiments. Like these I-V curves.
At this point in time, James was finally done with his thesis. He took the helm of our side of the project again, and he made himself very busy - I may even have to use the word "obsessed" - over the next few months...
He collected all I-V curves, he analysed GBs and GBs of data, and he put it all together in truly relentless grind, continuously cross-checking and optimising his Funtime Engine.
Now the work he and Tom performed on the STM paid off handsomely. As I mentioned, they worked on a complete rebuild/recode of our flagship instrument The Leviathan (now in its Mk.III
variant and currently undergoing some further changes towards Mk.IV), with the key objective of integrating a PID feedback loop to ensure a completely automatic/unsupervised data collection.
A "press-start-and-forget" approach. In essence, they were tired of babysitting the STM, so they wrote an algorithm that did it for them. Experiments that used to take a full day or even
multiple days now only take a few hours, which means we can quickly explore a wider range of experimental parameters, and easily do very demanding experiments. Like these I-V curves.
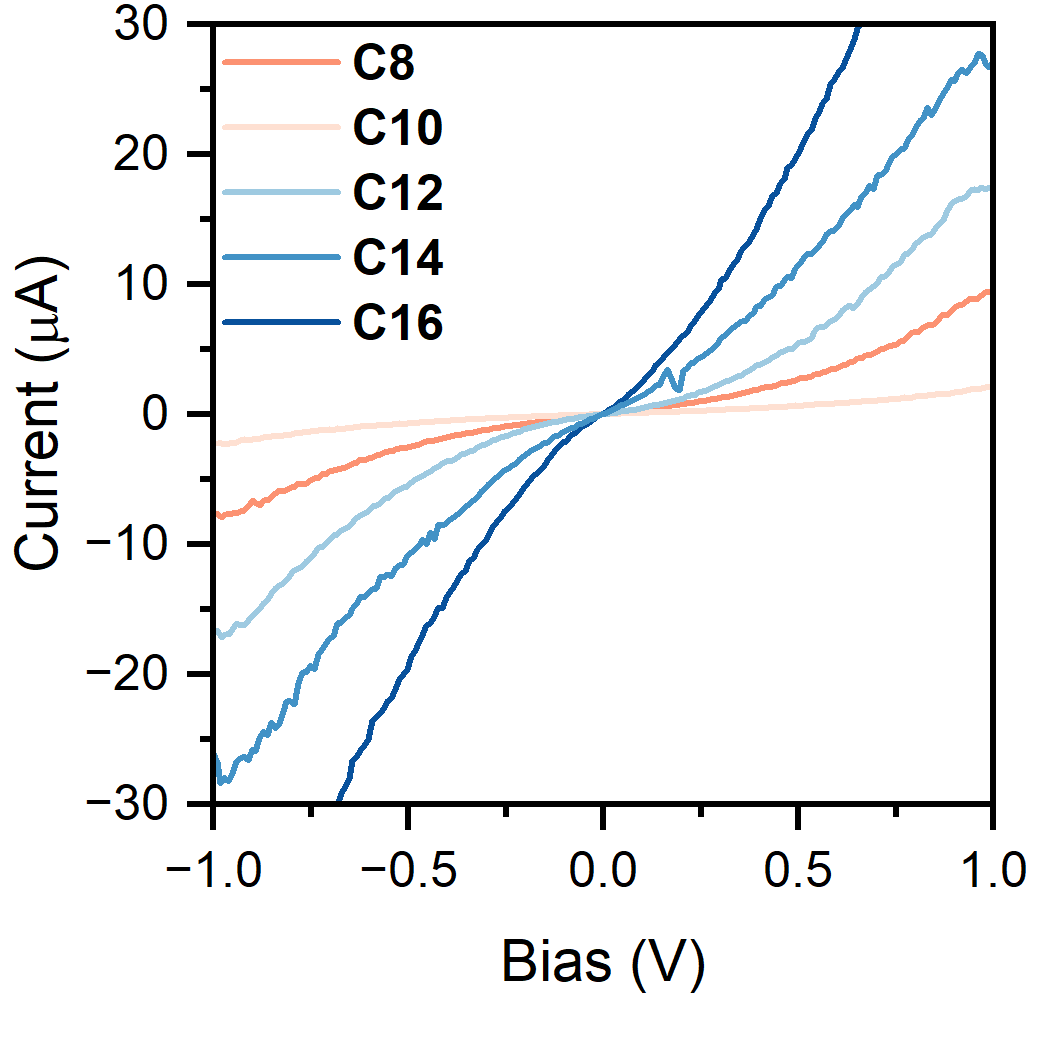 The data James (who also became Dr. Morris around this time) collected was an absolute bombshell.
As we go from C8 to C16 the evolution of the behaviour is stunningly clear: at low bias, the I-V curves mirrored perfectly the "static bias" conductance - decreasing from
C8 to C10, shooting up in C12 and then hovering around a fixed value. At higher bias however, data was unbelievable: the current through the carbon chain kept
increasing and increasing, reaching incredible values of ~40 μA in the longest derivative... You can see for yourself in the curves here on the side.
While James was busy with the I-V curves, Jarred also kept his hands occupied, trying to make C18 (!!) and C20 (!!!!) - which unfortunately did not work too well. C18 proved
impossible to purify to a level sufficient for our STMBJ experiments, while C20 - in Jarred's own words - "went up in flames on the sinter" (remember when I said to get behind the
blast shield? I wasn't joking). Nature loves reminding you that 1D carbon chains are essentially little sticks of
dynamite waiting for an excuse to return to soot. Nevertheless, we had already found the "Goldilocks zone": short enough to not go kaboom, but long enough to see the
quasi-ballistic cumulene-like behavior.
The data James (who also became Dr. Morris around this time) collected was an absolute bombshell.
As we go from C8 to C16 the evolution of the behaviour is stunningly clear: at low bias, the I-V curves mirrored perfectly the "static bias" conductance - decreasing from
C8 to C10, shooting up in C12 and then hovering around a fixed value. At higher bias however, data was unbelievable: the current through the carbon chain kept
increasing and increasing, reaching incredible values of ~40 μA in the longest derivative... You can see for yourself in the curves here on the side.
While James was busy with the I-V curves, Jarred also kept his hands occupied, trying to make C18 (!!) and C20 (!!!!) - which unfortunately did not work too well. C18 proved
impossible to purify to a level sufficient for our STMBJ experiments, while C20 - in Jarred's own words - "went up in flames on the sinter" (remember when I said to get behind the
blast shield? I wasn't joking). Nature loves reminding you that 1D carbon chains are essentially little sticks of
dynamite waiting for an excuse to return to soot. Nevertheless, we had already found the "Goldilocks zone": short enough to not go kaboom, but long enough to see the
quasi-ballistic cumulene-like behavior.
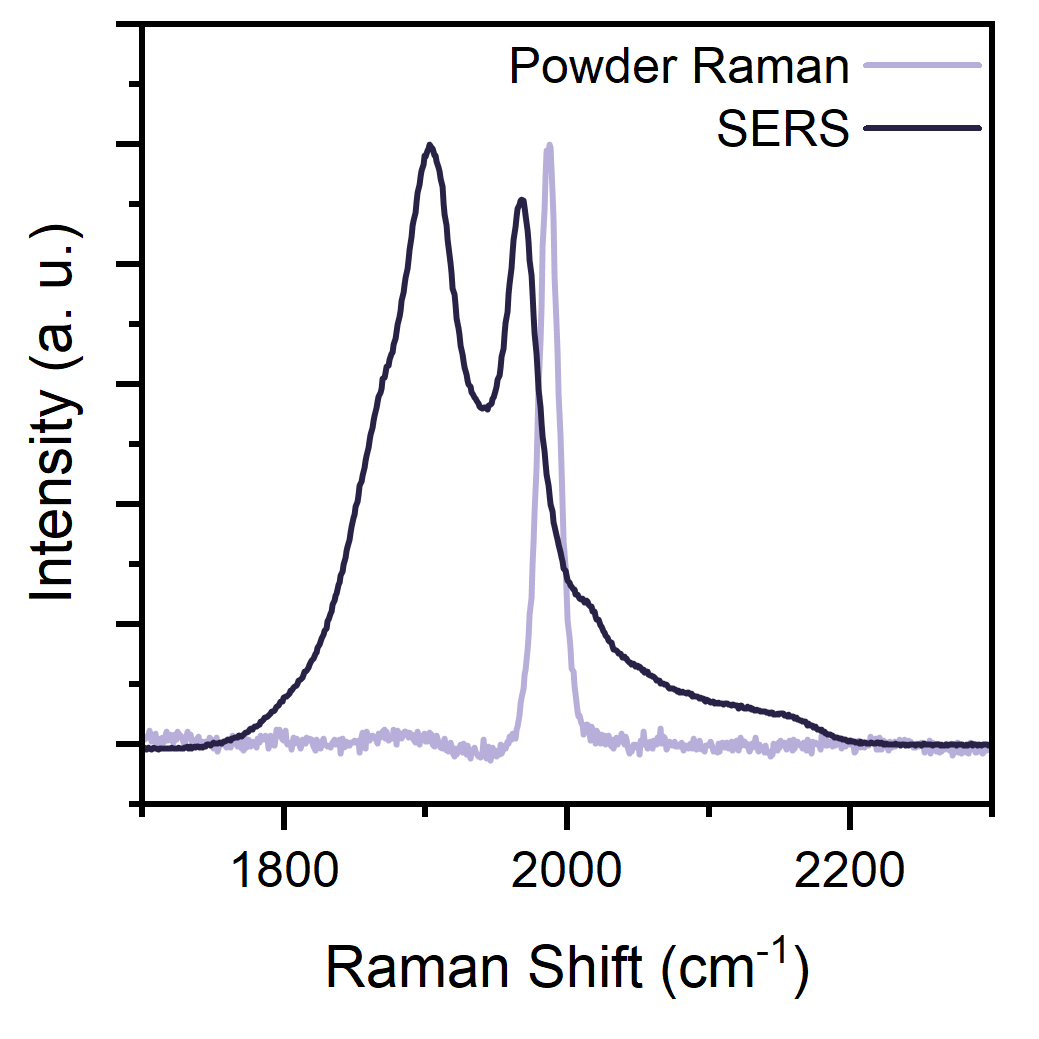 The last thing left to do was an independent proof of electronic equalisation. To get this, we turned our attention to Raman spectroscopy. Raman is ideal for
these carbon materials, as cumulenes and polyynes both have a very strong mode (the ECC - Effective Conjugation Coordinate - mode), which is the collective, in-phase stretching of all
carbon bonds. Key here is that if the structure is ...C≡C-C≡C... the ECC falls around 2000-2200 cm-1 while if it's ...C=C=C=C... the mode is redshifted towards 1800-1900
cm-1 - it's a sort of spectroscopic ruler for the BLA. To simulate the electrodes, we turned our attention to Au nanoparticles transmetallated to the carbon chain, exploiting their plasmonic
properties to further amplify the Raman signal and acquire extremely clean spectra. So - again - BACK TO THE LAB!! James made very nice "naked" nanoparticles, optimising a method we had already used
- with varying degrees of success - but this time purifying all materials to an insane standard and using only shiny new glassware. This obsession for perfection gave us absolutely pristine spectra, and guess what?
C16 behaved like a textbook example! The isolated material as Au(I) complex has the ECC at 2000 cm-1,
but it gets split and redshifted to 1900 cm-1 once it contacts the nanoparticles. We couldn't have asked for a better proof.
The last thing left to do was an independent proof of electronic equalisation. To get this, we turned our attention to Raman spectroscopy. Raman is ideal for
these carbon materials, as cumulenes and polyynes both have a very strong mode (the ECC - Effective Conjugation Coordinate - mode), which is the collective, in-phase stretching of all
carbon bonds. Key here is that if the structure is ...C≡C-C≡C... the ECC falls around 2000-2200 cm-1 while if it's ...C=C=C=C... the mode is redshifted towards 1800-1900
cm-1 - it's a sort of spectroscopic ruler for the BLA. To simulate the electrodes, we turned our attention to Au nanoparticles transmetallated to the carbon chain, exploiting their plasmonic
properties to further amplify the Raman signal and acquire extremely clean spectra. So - again - BACK TO THE LAB!! James made very nice "naked" nanoparticles, optimising a method we had already used
- with varying degrees of success - but this time purifying all materials to an insane standard and using only shiny new glassware. This obsession for perfection gave us absolutely pristine spectra, and guess what?
C16 behaved like a textbook example! The isolated material as Au(I) complex has the ECC at 2000 cm-1,
but it gets split and redshifted to 1900 cm-1 once it contacts the nanoparticles. We couldn't have asked for a better proof.
The "million dollar question" reamaining is the old one: why? It took us a while, but we have a good theory, backed by data. As the carbon chain grows, it becomes more electron-accepting. It craves electrons. This is quite clear in the NMR - the position of the resonance is dependent on the electrostatic environment, and the signals become more and more deshielded: the hallmark of electron-accepting behaviour. At the critical C12 length, they start to pull electrons out of the Au electrodes, through a phenomenon called d-π* back-donation: the d orbitals of Au have a good overlap with the antibonding π orbital of the terminal carbon - a good "conduit" for electrons. Once the antibonding orbital is populated, the corresponding bonding orbital is destabilised: the C≡C bond loses some π character, becoming more like a C=C bond. The extra electron density is given to the neighbouring C-C single bonds, which in turn gain some C=C character. In the end, all bonds are equalised towards the cumulene valence structure ...C=C=C=C... - all driven by this minute "backdonation" charge transfer between the molecule and the electrodes, like an electronic riptide. Does it make sense?
As usual, it all feels very anticlimactic in the end... the joy of the discovery is blurred and somewhat diluted by the sheer exhaustion of the verification and publication process. But that's science, right? Five minutes of excitement mean five months of tedious data collection, boring routine work to exhaust all alternative possibilities and rule out all alternative explanations, and - once the dust has settled - we even have to lay our hearts bare to editors and reviewers. And wait. Speaking for myself - and I guess also for 99% of all scientists in the world, since we're all still doing our experiments - those five minutes are absolutely worth it. For those five minutes, we have that wonderful feeling that we have pushed - even just by a fraction of a millimeter - the boundaries of human knowledge. It's the joy of realising that, after months and months of asking the same question over and over, Nature has finally whispered back the answer. I am incredibly grateful to Paul, James, Jarred and all our co-workers (Elena, Tom, Masnun, Chiara, Roberto, Eloise, Amit, Élodie, Simon, and Richard - as they say, it takes a village...) for giving me the opportunity to participate in this work. It's been an absolute tour de force, but I'd start again tomorrow. Or maybe the day after.
 Anyway, just to try to make the science behind all this a bit clearer and visually appealing, I instructed Gemini3 to produce some AI slop to help me.
You can adjust chain length in the below through the slider, and the structure will change from oligoyndiyl to cumulenic valence (it may not work correctly on mobile devices, I'm working on this).
The junction itself can be zoomed in and rotated. It's kinda pretty slop.
Anyway, just to try to make the science behind all this a bit clearer and visually appealing, I instructed Gemini3 to produce some AI slop to help me.
You can adjust chain length in the below through the slider, and the structure will change from oligoyndiyl to cumulenic valence (it may not work correctly on mobile devices, I'm working on this).
The junction itself can be zoomed in and rotated. It's kinda pretty slop.
Ah! Almost forgot... We've actually seen Jarred (or, more accurately, Dr. Potter) now. He's not a unicorn (proof attached) but - as James said - "just a bloke". I stand corrected.
I should make it absolutely clear here that this is my own personal account, and you will be able to read James and Jarred's perspective on the official SpringerNature website once it's published (Link TBA).
Edited 19/06/2026 to fix a few typos, mistakes, broken sentences and general sloppy writing. I also added the Leviathan official icon.
10/02/2025
Zero-Bias Anti-Ohmic Behaviour in Diradicaloid Molecular Wires
With the new term in full swing now, I oddly find myself with a bit more time on my hands. All lectures have been prepared, my physical chemistry module is working well,
the laboratory now keeps going without me (I'm more a handyman than a scientist lately, I only go in the lab to fix broken things) so I was getting ready to do some catch-up
with the literature when this email landed in my inbox:

 This is the sort of wake-up call that reminds me I should write another one of these "behind the paper" columns,
and in any case emails like this really make my day. So let's bite the bullet and dive in one of our
best publications of 2024, the Zero-Bias Anti-Ohmic Behaviour in Diradicaloid Molecular Wires" article we published in Angewandte Chemie. Buckle up kids, we're going through a long story with
lots of failures, disappointment, excitement, the worst nightmare for an academic, a trip across the pond and a final sprint to publication.
I had to scour my emails & Teams messages to reconstruct a sensible timeline, but I think this work started around May 2022. Amit (pictured here
smiling in the lab as he didn't know yet what he had signed up for) was working on the binaphthyl molecular wires we then published
here and he found out he could potentially make some quite interesting materials with just a few extra steps from
common precursors. He sent over a powerpoint slide with this diagram:
This is the sort of wake-up call that reminds me I should write another one of these "behind the paper" columns,
and in any case emails like this really make my day. So let's bite the bullet and dive in one of our
best publications of 2024, the Zero-Bias Anti-Ohmic Behaviour in Diradicaloid Molecular Wires" article we published in Angewandte Chemie. Buckle up kids, we're going through a long story with
lots of failures, disappointment, excitement, the worst nightmare for an academic, a trip across the pond and a final sprint to publication.
I had to scour my emails & Teams messages to reconstruct a sensible timeline, but I think this work started around May 2022. Amit (pictured here
smiling in the lab as he didn't know yet what he had signed up for) was working on the binaphthyl molecular wires we then published
here and he found out he could potentially make some quite interesting materials with just a few extra steps from
common precursors. He sent over a powerpoint slide with this diagram:

You can see the route to the binaphthyl derivative on top and its diversion to a bis(indeno)perylene from the triflate. Now that material seemed quite interesting - the reference providing the synthetic pathway to it shows it as having an interesting optoelectronic properties, so I started reading about these compounds, falling into the proverbial rabbit hole.
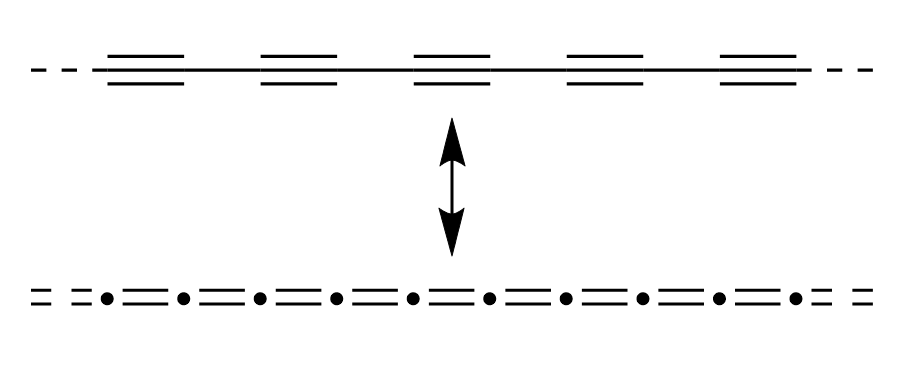
 That compound is an excellent example of a possible diradicaloid, a material which electronic structure
is best described by a superposition of canonical structures having different degrees of aromatic/close-shell and radical/open-shell character. I realise this sentence
is extemely difficult to parse, so I hope the figure on the side of this paragraph helps understanding it better. These compounds have a very odd electronic structure that can't be really shown
with a single Lewis-type structure. We can think of two possible "extreme" structures: the antiaromatic form, having no unpaired electrons but very
energetically unfavorable 4n electrons in the π-system, and an aromatic form, having two unpaired electrons (blasphemy!!) but a π-system with 4n+2 electrons, thus
having some degree of aromatic stabilisation. Both canonical structures are
high-energy so the "real" structure - the resonance hybrid - is something in-between these two extremes.
That compound is an excellent example of a possible diradicaloid, a material which electronic structure
is best described by a superposition of canonical structures having different degrees of aromatic/close-shell and radical/open-shell character. I realise this sentence
is extemely difficult to parse, so I hope the figure on the side of this paragraph helps understanding it better. These compounds have a very odd electronic structure that can't be really shown
with a single Lewis-type structure. We can think of two possible "extreme" structures: the antiaromatic form, having no unpaired electrons but very
energetically unfavorable 4n electrons in the π-system, and an aromatic form, having two unpaired electrons (blasphemy!!) but a π-system with 4n+2 electrons, thus
having some degree of aromatic stabilisation. Both canonical structures are
high-energy so the "real" structure - the resonance hybrid - is something in-between these two extremes.
 Now, this unusual electronic configuration gives this class of compounds many interesting properties.
Diradicaloids share many of the unusual features of radicals (see Saman's behind the paper
for some discussion) like high extinction coefficients granting vibrant and intense colours, enhanced reactivity and tendency to dimerise, and low-energy absorption giving them colours generally
not seen in organic materials, like deep green, blue or purple. The antiaromatic resonance tames reactivity a bit, and makes these materials generally more easy to manipulate
than proper radicals, even in air or oxidising environment, as long as some thought is given during the synthetic process to ensure that the position of high spin density (those part of the molecule
where the unpaired electrons will most likely localise) are sufficiently protected. Many research groups have made huge contributions to the understanding of structure-property relationships
in these materials, and we had an incredibly vast chemical space to explore to design our molecular wires. To us, these systems looked like the perfect candidate to start introducing multiple unpaired electrons,
and see how their presence would affect charge transport. Their syntheses can however be quite tedious and time consuming, so we had to thread lightly and
try to find the best direction for our efforts. After reading many papers and reviews - and the fantastic book "Polyarenes I" with a chapter by
Michael Haley from the University of Oregon dedicated to diradicaloid compounds -
we settled on investigating a small group of these materials, based on the [1,2-b]indenofluorene scaffold, and try to extend the acene system to explore how conductance evolves in materials
of increasing length. Our initial choice of targets was very simple - something like the ones here - using thioanisole anchors to the electrodes, two methyls on the thioanisole to twist
the aromatic ring out of conjugation and protect the radical states, and an acene of increasing length between the two indenyl groups. There are routes to all these but there's a catch: while the indenofluorene
(the shortest compound) is relatively easy to make, the other two are not: the naphthyl and the anthryl derivatives are not exceedingly stable, and their syntheses are complicated by steps that
can give multiple isomers difficult to isolate, poorly soluble intermediates, and generally unfriendly chemistry.
Now, this unusual electronic configuration gives this class of compounds many interesting properties.
Diradicaloids share many of the unusual features of radicals (see Saman's behind the paper
for some discussion) like high extinction coefficients granting vibrant and intense colours, enhanced reactivity and tendency to dimerise, and low-energy absorption giving them colours generally
not seen in organic materials, like deep green, blue or purple. The antiaromatic resonance tames reactivity a bit, and makes these materials generally more easy to manipulate
than proper radicals, even in air or oxidising environment, as long as some thought is given during the synthetic process to ensure that the position of high spin density (those part of the molecule
where the unpaired electrons will most likely localise) are sufficiently protected. Many research groups have made huge contributions to the understanding of structure-property relationships
in these materials, and we had an incredibly vast chemical space to explore to design our molecular wires. To us, these systems looked like the perfect candidate to start introducing multiple unpaired electrons,
and see how their presence would affect charge transport. Their syntheses can however be quite tedious and time consuming, so we had to thread lightly and
try to find the best direction for our efforts. After reading many papers and reviews - and the fantastic book "Polyarenes I" with a chapter by
Michael Haley from the University of Oregon dedicated to diradicaloid compounds -
we settled on investigating a small group of these materials, based on the [1,2-b]indenofluorene scaffold, and try to extend the acene system to explore how conductance evolves in materials
of increasing length. Our initial choice of targets was very simple - something like the ones here - using thioanisole anchors to the electrodes, two methyls on the thioanisole to twist
the aromatic ring out of conjugation and protect the radical states, and an acene of increasing length between the two indenyl groups. There are routes to all these but there's a catch: while the indenofluorene
(the shortest compound) is relatively easy to make, the other two are not: the naphthyl and the anthryl derivatives are not exceedingly stable, and their syntheses are complicated by steps that
can give multiple isomers difficult to isolate, poorly soluble intermediates, and generally unfriendly chemistry.
 Nevertheless, Amit bravely started to work on these materials, and by August 2022 he had his hands on an analytically pure sample
of the shortest derivative in the series - see here on the left the key final oxidation that stripped two protons from the orange-ish precursor to give the lovely, deep purple indenofluorene.
We tested its behaviour in single-molecule junctions and we could see straight away that it was an interesting compound - while low-bias conductance was relatively low, it rapidly increased with increasing bias.
These non-linear current-voltage behaviour is the hallmark of unusual electronic characteristics, and in our field it usually means that the transport orbital is
close in energy to the Fermi level of the electrodes.
Such phenomena are not common, and they're quite interesting to study. We really wanted now to make the longer ones, on which we ran some initial calculations and seemed to hold so much promise.
Well, making these turned out to be even more complicated than originally expected.
Nevertheless, Amit bravely started to work on these materials, and by August 2022 he had his hands on an analytically pure sample
of the shortest derivative in the series - see here on the left the key final oxidation that stripped two protons from the orange-ish precursor to give the lovely, deep purple indenofluorene.
We tested its behaviour in single-molecule junctions and we could see straight away that it was an interesting compound - while low-bias conductance was relatively low, it rapidly increased with increasing bias.
These non-linear current-voltage behaviour is the hallmark of unusual electronic characteristics, and in our field it usually means that the transport orbital is
close in energy to the Fermi level of the electrodes.
Such phenomena are not common, and they're quite interesting to study. We really wanted now to make the longer ones, on which we ran some initial calculations and seemed to hold so much promise.
Well, making these turned out to be even more complicated than originally expected.

 We originally tried to make these going through the
synthesis of the corresponding biscarbonyl precursor. There are published routes to all these, but we couldn't isolate them in sufficient purity
to continue our work - the dione is made by oxidative ring closure, and it can close in many positions, giving a statistical distribution
of the possible products (see here on the right, in orange). And those obviously
proved impossible to separate from each other, mostly because of their low solubility. So we had to change our strategy a bit, and start introducing some substituents on the acene
to try to tame its reactivity and direct it to the desired compound. James Burrows, a brilliant undergraduate working in our
laboratories as part of his 3rd year project, initially tested a tetrahydropyrene derivative
(the thing in green here) that actually delivered the desired compound. Everything seemed to be working well
(although the synthesis itself sometimes yielded some monsters like the one pictured here below the structures)
and we had a sample by the end of his project. While the compound seemed the correct one from its spectroscopic signature, it did not work well in the STMBJ - any attempt at fabricating
single-molecule junctions returned very low and ill-defined conductance - especially when biases >300 mV were used - and in general it performed very poorly.
Our attempts at growing crystals of this material for SCXRD characterisation also kept giving us mostly black tar and teeny-tiny crystals of the reduction product:
this compound was not stable and most probably we were observing decomposition when assembled as single-molecule junction.
Time to go back to the drawing board and try to understand what's happening here.
We originally tried to make these going through the
synthesis of the corresponding biscarbonyl precursor. There are published routes to all these, but we couldn't isolate them in sufficient purity
to continue our work - the dione is made by oxidative ring closure, and it can close in many positions, giving a statistical distribution
of the possible products (see here on the right, in orange). And those obviously
proved impossible to separate from each other, mostly because of their low solubility. So we had to change our strategy a bit, and start introducing some substituents on the acene
to try to tame its reactivity and direct it to the desired compound. James Burrows, a brilliant undergraduate working in our
laboratories as part of his 3rd year project, initially tested a tetrahydropyrene derivative
(the thing in green here) that actually delivered the desired compound. Everything seemed to be working well
(although the synthesis itself sometimes yielded some monsters like the one pictured here below the structures)
and we had a sample by the end of his project. While the compound seemed the correct one from its spectroscopic signature, it did not work well in the STMBJ - any attempt at fabricating
single-molecule junctions returned very low and ill-defined conductance - especially when biases >300 mV were used - and in general it performed very poorly.
Our attempts at growing crystals of this material for SCXRD characterisation also kept giving us mostly black tar and teeny-tiny crystals of the reduction product:
this compound was not stable and most probably we were observing decomposition when assembled as single-molecule junction.
Time to go back to the drawing board and try to understand what's happening here.

 It took us a long time to figure it out, but we
eventually realised we had issues with aerobic oxidation. All acenes suffer from it, by reaction with oxygen and light to given an endoperoxide that then further reacts irreversibly
leaving only a disgusting, sticky tar-like residue. Anthryl derivatives are usually fine, but in our case the lateral π-extension was giving problems.
Luckily, there are strategies to mitigate decomposition, for instance by protecting the acene with ethynyl substituents.
Rather than preventing oxidation itself, the ethynyl substituents promote fast and clean thermolytic reconversion of the endoperoxide back to the acene - and interestingly, we found out that
Haley and co-workers used this exact strategy to stabilise a bis(indeno)anthryl derivative! With this knowledge, it was easy to design the
final compounds we needed for our study - although it took us quite some time get here! Amit started working on these straight away and whipped out the anthryl derivative in a
couple of months. What's beautiful there is that the ethynyl substituents not only provide protection agains oxidation, but they also add enough steric bulk to direct the ring-closure reaction
to the desired product - the proverbial two-birds-with-a-stone!
It took us a long time to figure it out, but we
eventually realised we had issues with aerobic oxidation. All acenes suffer from it, by reaction with oxygen and light to given an endoperoxide that then further reacts irreversibly
leaving only a disgusting, sticky tar-like residue. Anthryl derivatives are usually fine, but in our case the lateral π-extension was giving problems.
Luckily, there are strategies to mitigate decomposition, for instance by protecting the acene with ethynyl substituents.
Rather than preventing oxidation itself, the ethynyl substituents promote fast and clean thermolytic reconversion of the endoperoxide back to the acene - and interestingly, we found out that
Haley and co-workers used this exact strategy to stabilise a bis(indeno)anthryl derivative! With this knowledge, it was easy to design the
final compounds we needed for our study - although it took us quite some time get here! Amit started working on these straight away and whipped out the anthryl derivative in a
couple of months. What's beautiful there is that the ethynyl substituents not only provide protection agains oxidation, but they also add enough steric bulk to direct the ring-closure reaction
to the desired product - the proverbial two-birds-with-a-stone!
 As soon as we started our measurements, we could see they were delivering the expected results.
The anthryl derivative was more conductive than the shortest indenofluorene,
and its conductance increased even more rapidly with bias! Making the naphthyl analogue proved to be more of a challenge, but we eventually managed to get there - and from the
timeline I've been able to reconstruct (this time helped by the timestamp on our measurements) this puts us around the end of 2023. Since we finally had a good set of compounds,
delivering very interesting results, we started working to ensure our results were reproducible, make our statistics more robust, growing all crystals for XRD measurements,
re-make a couple of intermediates with substandard characterisation, etc. - and in the meanwhile, our theoretical collaborators in Warwick were busy with their calculations to model
the behaviour we observed. Lots of back-and-forth between the two of us pointed us to a possible explanation for the observed behaviour. These materials are singlet diradicaloids:
antiferromagnetic coupling between the two unpaired electrons forces them to have opposite spins.
As soon as we started our measurements, we could see they were delivering the expected results.
The anthryl derivative was more conductive than the shortest indenofluorene,
and its conductance increased even more rapidly with bias! Making the naphthyl analogue proved to be more of a challenge, but we eventually managed to get there - and from the
timeline I've been able to reconstruct (this time helped by the timestamp on our measurements) this puts us around the end of 2023. Since we finally had a good set of compounds,
delivering very interesting results, we started working to ensure our results were reproducible, make our statistics more robust, growing all crystals for XRD measurements,
re-make a couple of intermediates with substandard characterisation, etc. - and in the meanwhile, our theoretical collaborators in Warwick were busy with their calculations to model
the behaviour we observed. Lots of back-and-forth between the two of us pointed us to a possible explanation for the observed behaviour. These materials are singlet diradicaloids:
antiferromagnetic coupling between the two unpaired electrons forces them to have opposite spins.  By having opposite spin, they create new transport resonances (sort of degenerate SOMO/SUMO) separated by a "transport gap", within the classical, close-shell
HOMO-LUMO gap, spanning the entire molecular backbone - and very similar in wavefunction to the spin-density map we used as guideline for our synthetic design. Most important, the new
transport gap shrinks with increasing acene length at an incredibly rapid pace, aided by the increase in diradicaloid character of these compound - so by the greater ability of longer
diradicaloids in efficiently delocalising the unpaired electrons. While long molecular wires tend to self-decouple from the electrodes, thus becoming more resistive, in this case the transport
gap shrinks at such a rate that it overcomes the natural self-decoupling, giving us the observed anti-ohmic behaviour. Most importantly, the effect
is consistent across the entire bias window, returning massive values of conductance increase - a negative β-factor - at high voltages (>1 V).
By having opposite spin, they create new transport resonances (sort of degenerate SOMO/SUMO) separated by a "transport gap", within the classical, close-shell
HOMO-LUMO gap, spanning the entire molecular backbone - and very similar in wavefunction to the spin-density map we used as guideline for our synthetic design. Most important, the new
transport gap shrinks with increasing acene length at an incredibly rapid pace, aided by the increase in diradicaloid character of these compound - so by the greater ability of longer
diradicaloids in efficiently delocalising the unpaired electrons. While long molecular wires tend to self-decouple from the electrodes, thus becoming more resistive, in this case the transport
gap shrinks at such a rate that it overcomes the natural self-decoupling, giving us the observed anti-ohmic behaviour. Most importantly, the effect
is consistent across the entire bias window, returning massive values of conductance increase - a negative β-factor - at high voltages (>1 V).
Now, credit where it's due: we didn't invent anything new. Thijs Stuyver explored these concepts theoretically in 2018, building on previous work performed in collaboration with none other than Roald Hoffmann, so what we did was really just take these concepts and prove them in the lab, in two gruelling years of toil, tears and sweat (no blood this time, I can guarantee no students or postdocs were harmed during this study). Anyway, now we had everything we needed, so we started writing this up for publication, while the spring meeting of the American Chemical Society appeared on the horizon. Our idea was to present this data there, collect some feedback from colleagues, and then come back to Liverpool, finish the paper, and submit. A couple of days before leaving for New Orleans, however, I woke up to this email notifying me of a new preprint on the arXiv citing our past work on verdazyl radicals:

I still remember I started to have cold sweats - we were being scooped!! Maria Kamenetska and Michael Haley (I guess you've seen his name enough times now to know that he's one of the eminences in diradicaloid chemistry) have been working on the same compounds we originally targeted, and they managed to make them! It was quite a bummer - and I remember seeing Amit particularly dispirited - but after carefully reading their preprint, I noticed they seemed to have had some of the same issues we encountered. The naphthyl and anthryl compounds proved to be not stable enough, and they could only characterise them at low voltages (~250 mV) - while our materials were stable to biases of >1.5 V. It wasn't exactly what I hoped, but I could see a way for both our studies to be relevant and interesting, and to survive the conference without looking like an absolute fool. A quick look at the schedule let me realise I was supposed to be speaking shortly after Maria, so I decided to bin the presentation I prepared and write a new one from scratch, focussing on the strategy we employed to stabilise these materials to withstand strong voltages and electric fields rather than on the results. All considered, I was in a relatively lucky position, and I had plenty of time on my 8-hour transatlantic flight to whip up a good presentation. The conference was a blast, and all went pretty well in the end.
We followed the proposed timeline and I uploaded a preprint on ChemRxiv upon returning to the UK, followed by enrolling our manuscript in the submission roulette. In a couple of months, our work was published in Angewandte Chemie, and Maria's study instead found a cozy home in ACS Nano. Quite interestingly, just a few months after our respective works were published, another study exploiting the diradicaloid behaviour of indenofluorene derivatives found its way into the literature - this time by Cuerva, Leary and Millan. I guess this last part shows that these materials are contributing a lot to our understanding of charge transport through molecular junctions, and I can safely bet this won't be the last time we see radicals and diradicaloids used in our field, with excellent results.
 I guess that's all for this paper - I hope you enjoyed the ride. I want to use this space to thank
all people who contributed to our study: Amit, the superstar of this paper (just take a minute to admire the massive needle-like crystals of the naphthyl derivative here on the left), who was aided
by the two James (Burrows and Morris) in, respectively, the synthetic work and the single-molecule junction characterisation. Lewis and Abdul for their work on the theory, Konstantin our
resident NMR wizard for his help with the VT-NMR one of the reviewer requested as part of our revision, and Craig the master crystallographer for getting excellently resolved structures out of our
teeny-tiny crystals. And obviously, last round of thanks to all my long-standing collaborators - Richard, Simon, Hatef & Sara - for the excellent work.
I guess that's all for this paper - I hope you enjoyed the ride. I want to use this space to thank
all people who contributed to our study: Amit, the superstar of this paper (just take a minute to admire the massive needle-like crystals of the naphthyl derivative here on the left), who was aided
by the two James (Burrows and Morris) in, respectively, the synthetic work and the single-molecule junction characterisation. Lewis and Abdul for their work on the theory, Konstantin our
resident NMR wizard for his help with the VT-NMR one of the reviewer requested as part of our revision, and Craig the master crystallographer for getting excellently resolved structures out of our
teeny-tiny crystals. And obviously, last round of thanks to all my long-standing collaborators - Richard, Simon, Hatef & Sara - for the excellent work.
I know this won't work, but I'll try anyway: this work will be continued and I wish to reserve the field for myself (cit.)
Edited 11/02/25 to fix a few typos and general polish.
Re-edited 19/02/25 to fix another couple of typos.
Re-re-edited 25/02/2025 because it looks like I can't tell the difference between left and right.
17/04/2023
Redox-Addressable Single-Molecule Junctions Incorporating a Persistent Organic Radical
I’ve just noticed it’s quite a long time I haven’t written one of these… But I must admit the last couple of years I’ve been quite busy
(and I have some great news lately so all this being busy somehow paid off)
so maybe I haven’t had the time (or the willingness) to sit down and write. But now I can relax and finally celebrate
this paper – one that has been a long and winding road,
full of many little pieces that had to fall into place before we could move along.
 This time, everything started with a parcel in my pigeon
hole. Varshini and Paul from UWA Australia sent on a
15000 km trip a few vials, containing some compounds with a strange structure, something I wasn’t actually familiar with.
The cover letter stated something like “please also find included some 6-oxoverdazyl derivatives and tetrazane precursors we’ve recently
synthesised – let us know if they’re interesting”.
Well, the first thing I did was to google “verdazyl” to try to educate myself. And it turns out
verdazyls are amongst the most stable organic radicals known to man.
This time, everything started with a parcel in my pigeon
hole. Varshini and Paul from UWA Australia sent on a
15000 km trip a few vials, containing some compounds with a strange structure, something I wasn’t actually familiar with.
The cover letter stated something like “please also find included some 6-oxoverdazyl derivatives and tetrazane precursors we’ve recently
synthesised – let us know if they’re interesting”.
Well, the first thing I did was to google “verdazyl” to try to educate myself. And it turns out
verdazyls are amongst the most stable organic radicals known to man.
That was indeed quite interesting, and I fell down the rabbit hole.
 Organic radicals are insanely fascinating. They’re species with unpaired electrons (open-shell), and as such
they are usually extremely reactive - electrons definitely don’t like being alone! There are however ways (“a combination of sterics and electronics!”) to make radicals
sufficiently stable that they can be isolated. The story of organic radicals starts in 1900, with Gomberg’s successful synthesis of the triphenylmethyl
(trityl) radical. The paper by Gomberg (his face here on the left) is a work of art by itself, and it ends with the amazing sentence “this work will be continued and I wish to reserve
the field for myself”. Well, thankfully nobody respected Gomberg’s wish, and since then several other organic radicals have been synthesised and characterised.
Kuhn and Trischmann discovered in 1963 a “surprisingly stable nitrogenous free radical” (this is the actual title of the paper, also published in Angewandte Chemie!),
when they methylated a formazan derivative. The resulting compound spontaneously dehydrogenated in the presence of atmospheric oxygen to yield an open-shell species
with an intense green colour. The authors suggested the trivial name “verdazyl” for these compounds – my guess here is that it’s taken from my own language, Italian,
where green is “verde”, -az- comes from the nitrogen-based structure and -yl denotes the radical character. Kuhn and Trischmann tried to wreck this radical in many ways – they even
boiled it for long time in acetic acid or concentrated methoxide solution, but could observe no decomposition. Only treatment with concentrated hydrochloric acid promoted
dismutation to the deep purple radical cation and the neutral species – called leucoverdazyl as a nod to old-school dye chemistry. Anyway, they didn’t reserve the verdazyl
field for themselves, and since their report, verdazyls have been extensively functionalised and proposed for uses in a range of applications, from spin labels to components
of magnetic and conductive materials, as ligands for transition metal complexes and as redox components of organic-based batteries.
In our field – single-molecule electronics – the existence of an unpaired electron would impart to the molecular junctions several exciting properties.
Theoretical calculations predict, for instance, high efficiency in charge transport and thermoelectric conversion, and many attempts at
incorporating organic radicals in single-molecule junctions have been made in the past. Unfortunately, while evidence of persistence of the open-shell
(radical) state has been reported at cryogenic temperature, when we wrote this paper no such thing was reported at room temperature
(Now there's a report in Nano Letters about their enhanced thermoelectric properties).
As I said earlier, radicals are not stable, and in most cases charge transfer from the junction electrodes would quench the open-shell state, leaving behind
just a boring close-shell species with no interesting properties.
Organic radicals are insanely fascinating. They’re species with unpaired electrons (open-shell), and as such
they are usually extremely reactive - electrons definitely don’t like being alone! There are however ways (“a combination of sterics and electronics!”) to make radicals
sufficiently stable that they can be isolated. The story of organic radicals starts in 1900, with Gomberg’s successful synthesis of the triphenylmethyl
(trityl) radical. The paper by Gomberg (his face here on the left) is a work of art by itself, and it ends with the amazing sentence “this work will be continued and I wish to reserve
the field for myself”. Well, thankfully nobody respected Gomberg’s wish, and since then several other organic radicals have been synthesised and characterised.
Kuhn and Trischmann discovered in 1963 a “surprisingly stable nitrogenous free radical” (this is the actual title of the paper, also published in Angewandte Chemie!),
when they methylated a formazan derivative. The resulting compound spontaneously dehydrogenated in the presence of atmospheric oxygen to yield an open-shell species
with an intense green colour. The authors suggested the trivial name “verdazyl” for these compounds – my guess here is that it’s taken from my own language, Italian,
where green is “verde”, -az- comes from the nitrogen-based structure and -yl denotes the radical character. Kuhn and Trischmann tried to wreck this radical in many ways – they even
boiled it for long time in acetic acid or concentrated methoxide solution, but could observe no decomposition. Only treatment with concentrated hydrochloric acid promoted
dismutation to the deep purple radical cation and the neutral species – called leucoverdazyl as a nod to old-school dye chemistry. Anyway, they didn’t reserve the verdazyl
field for themselves, and since their report, verdazyls have been extensively functionalised and proposed for uses in a range of applications, from spin labels to components
of magnetic and conductive materials, as ligands for transition metal complexes and as redox components of organic-based batteries.
In our field – single-molecule electronics – the existence of an unpaired electron would impart to the molecular junctions several exciting properties.
Theoretical calculations predict, for instance, high efficiency in charge transport and thermoelectric conversion, and many attempts at
incorporating organic radicals in single-molecule junctions have been made in the past. Unfortunately, while evidence of persistence of the open-shell
(radical) state has been reported at cryogenic temperature, when we wrote this paper no such thing was reported at room temperature
(Now there's a report in Nano Letters about their enhanced thermoelectric properties).
As I said earlier, radicals are not stable, and in most cases charge transfer from the junction electrodes would quench the open-shell state, leaving behind
just a boring close-shell species with no interesting properties.
And now back to us.
After educating myself on verdazyl chemistry I understood Paul and Varshini’s choice. The unpaired electron is strongly localised on the 4 nitrogen
atoms, and the 6-oxoverdazyl derivatives they made have a particularly high oxidation potential. These two effects combined would give these compounds a
higher chance of surviving unscathed when sandwiched between two metallic electrodes, reducing charge-transfer induced redox processes that would quench
the open-shell state - an issue already encountered in other radicals. Time to fabricate some junctions!
 In this project, Saman took the leading role in the experiments in our lab. He had plenty of experience on the STMs and he was looking for a new
challenge: the verdazyls were right down his alley. He started by measuring the longer compounds, with the verdazyl core flanked by 4-ethynyl(pyridine)
or 4-ethynyl(thioanisole), and their closed-shell analogues (tetrazin-3-one). And here we hit the first wall. While we could measure conductance through
the open-shell species, the closed-shell one was too poorly conductive to be measured. So - we had some exciting results, but nothing to compare them with to confirm
retention of the radical state of the junction. We then suggested Paul and Varshini to try to make some shorter analogues, without ethynyl bridges. These "2nd generation compounds"
worked like a charm: the 6-oxoverdazyl had greatly enhanced charge transport propertis when compared to the tetrazin-3-one. We finally had a first, strong proof that the unpaired
electron was still on the molecular junction, but this wasn’t enough. We wanted to be sure we had the molecule trapped in the open-shell, radical state, and that no quenching
(self-immolation of the radical) was happening.
In this project, Saman took the leading role in the experiments in our lab. He had plenty of experience on the STMs and he was looking for a new
challenge: the verdazyls were right down his alley. He started by measuring the longer compounds, with the verdazyl core flanked by 4-ethynyl(pyridine)
or 4-ethynyl(thioanisole), and their closed-shell analogues (tetrazin-3-one). And here we hit the first wall. While we could measure conductance through
the open-shell species, the closed-shell one was too poorly conductive to be measured. So - we had some exciting results, but nothing to compare them with to confirm
retention of the radical state of the junction. We then suggested Paul and Varshini to try to make some shorter analogues, without ethynyl bridges. These "2nd generation compounds"
worked like a charm: the 6-oxoverdazyl had greatly enhanced charge transport propertis when compared to the tetrazin-3-one. We finally had a first, strong proof that the unpaired
electron was still on the molecular junction, but this wasn’t enough. We wanted to be sure we had the molecule trapped in the open-shell, radical state, and that no quenching
(self-immolation of the radical) was happening.
So we decided to quench the radical on purpose.
We did so by exploiting the rich electrochemistry of verdazyl radicals, which can be reduced or oxidised from the open-shell 7π electronic structure to, respectively, close-shell
8π anionic or 6π cationic state. As discussed earlier, open-shell materials are expected to have more efficient transport properties, and therefore we would expect a drop in conductance
as the molecule gets electrochemically oxidised or reduced. And since we had our nice electrochemical STM setup to use (see here for some further discussion on ECSTM),
we started collecting data under electrochemical control. Again, in ionic liquids. Which, as already discussed, are an absolute pain to purify. We bought a few bottles of
1-butyl-3-methylimidazolium triflate, heated them over molecular sieves until we had a wide enough electrochemical window, and then started fabricating junctions. A lot of them.
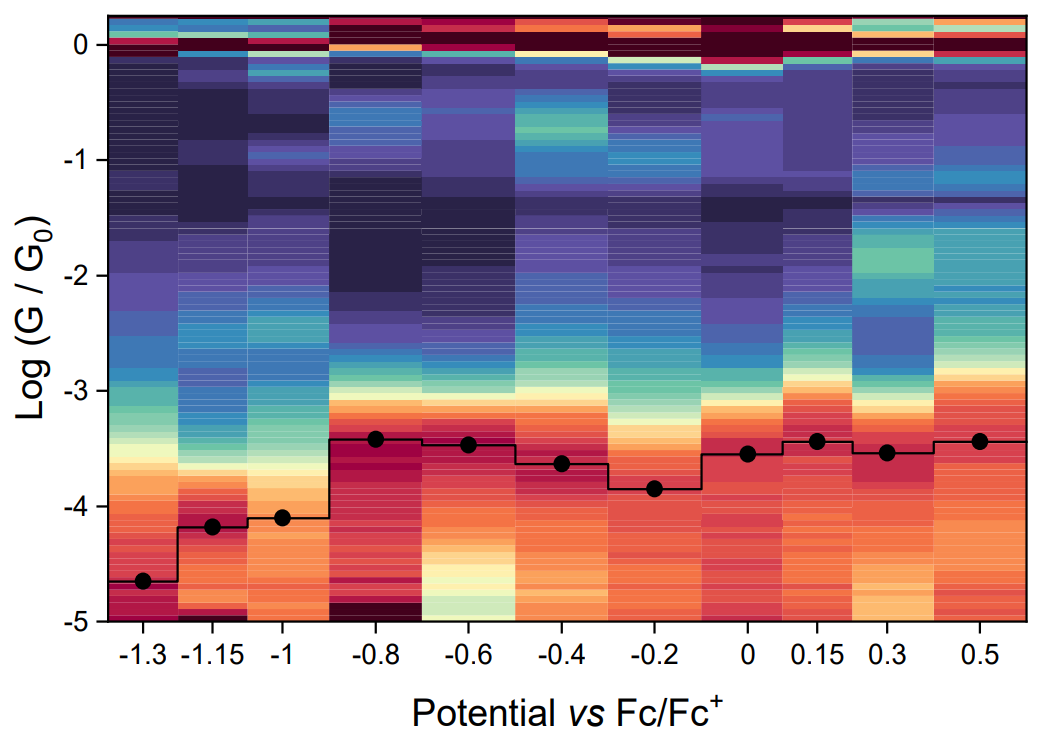 Saman collected data over a huge potential window, to try and access both the oxidised and reduced state - unfortunately our
equipment went ballistic at high positive potentials, so we could only characterise the 7π → 8π transition, but it was clear enough to confirm our expectations.
At the redox potential, when the verdazyl radical is reduced to the anionic state, conductance suddenly drops by almost an order of magnitude, a phenomenon we too as a hallmark of the quenching of the
open-shell state. We would expect a similar drop in conductance upon oxidation to the cationic state, and Saman tried several times to
obtain a measurement at high positive potentials, but it didn't work at all. Well, you can't always win.
Saman collected data over a huge potential window, to try and access both the oxidised and reduced state - unfortunately our
equipment went ballistic at high positive potentials, so we could only characterise the 7π → 8π transition, but it was clear enough to confirm our expectations.
At the redox potential, when the verdazyl radical is reduced to the anionic state, conductance suddenly drops by almost an order of magnitude, a phenomenon we too as a hallmark of the quenching of the
open-shell state. We would expect a similar drop in conductance upon oxidation to the cationic state, and Saman tried several times to
obtain a measurement at high positive potentials, but it didn't work at all. Well, you can't always win.
It was time to ask our trusted theoreticians to model the compounds and give us further insights on their behaviour. We asked
Sara Sangtarash and Hatef Sadeghi to help us out, and they performed a thorough modelling which improved
our understanding of the system. The radical has two additional orbitals, a semioccupied and a semi-unoccupied one (SOMO and SUMO) which sit at energies close to the Fermi level of the electrodes. While we can't directly
access them (calculations predict that the energy of the tunnelling electrons sits in the SOMO/SUMO gap) their presence raises the mid-bandgap transmission significantly. Our electrochemical gating allows us to see
this effect as a slight increase in conductance as the potential is moved away from -0.2 V vs. Fc/Fc+ in both directions. As the electrochemical potential reaches the energy of the SOMO or the SUMO,
the material is oxidised to a wide-bandgap species (the leuco form of the radical).
Now everything (almost) made sense, we were ready to submit this work for publication. We drafted a paper, went through lots of iterative optimisation steps, which required collecting some more data here and there, extra
quick calculations, etc. but we were pretty happy with the result. In this case, I believe we were pretty thorough in our investigation and, after peer-review, the comments we received were all about minor issues:
a horrible mess created by my reference manager, a few experimental details we duly added to the SI, and clarification on some language we used.
It must be said that this account covers only part of the work presented in the article - Saman also performed some I-V experiments to determine the current-voltage characteristics of these materials, and found a quite
funny behaviour. Our collaborators in Australia also performed EPR on the verdazyl compounds, even when adsorbed on Au substrates as monolayers, to check the existnce of the open-shell state even in the
presence of a metallic electrode, and you can read all about these in the manuscript (which is open-access and free to read). This "behind the paper"
is mostly a celebration of the hard work of the research teams involved, and Saman's refusal to give up even when faced with many things that didn't work - the first series of long compounds which gave
unsatisfactorily results, the constantly failing electrochemical measurements at high positive potentials and the general challenges of working with electrochemical STM and ionic liquids. Looking back, the amount
of data acquired is again quite staggering: ~75GB, corresponding to ~110000 single-molecule junctions fabricated. What can I say? We love BIG data!
Many thanks to all people involved in this study, and to Tom Abram for reminding me over and over that it was time to write another "behind the paper". I'll try not to wait another two years to write the next one.
Edited 17/06/26 because finally VS Code has a built-in spellchecker.
13/10/2020
Folding a Single-Molecule Junction
It’s always nice to have the opportunity to write another behind the paper, this time for an article we recently published in Nano Letters! In this case, everything started quite a long time ago – I think around summer 2016. Mr. Aidan Thomas was working on his summer project in Dr. Craig Robertson’s lab, that was just upstairs from my office at the time. He was working with Craig on a series of compounds akin to benzil (1,2-diphenylethane-1,2-dione) and for one of the syntheses, he had to use quite a decent amount of n-butyllithium in a synthetic process originally developed by Italian chemists (some pride here!). In those years, I was going through bottles and bottles of n-, sec- and t-butyllithium per month, so Craig asked me to help Aidan out with his work, and show him how to work with pyrophorics in a safe way. For the organikers interested in the methods, here’s the synthesis he was tasked with:
 Anyway, in late 2016 Craig’s new PhD student, (now soon-to-be-Dr.) Demetris Bates picked
up Aidan’s work, so I trained him in the use of pyrophorics. He ended up playing the leading synthetic role in this project, as you'll see later.
He is a fantastic chemist, so it took him little time to get to grips with the reaction, optimise its conditions and make a good batch of the compound
above (also in its crystalline form as massive yellow needles).
Demetris gave me a bottle with 20-30 mg of this compound as I thought it could allow the
fabrication of junctions with interesting behaviour. I took the bottle, put it in one of my desk drawers, and completely forgot about it. Yes. I am sorry Demetris,
I completely forgot. In my defence, in 2017 I was an over-caffeinated, sleep-deprived, stressed-out, new-dad PDRA, so some memory impairment was only natural.
More than 2 years later, the stars aligned. In early September 2019 I was finally given an office of my own, so I cleared out my desk – and voila’,
in the back of one of my drawers, I found the little sample bottle Demetris gave me. Chuanli (see here if you don’t have a clue who she is) had just
finished her work on the polyoxometalate molecular wire, and she wanted to have something to do in her last couple of weeks in the UK. Her visit to our
lab was supposed to end on the 30th of September 2019, so I gave her the bottle with Demetris' compound to start a few break-junction experiments. We could see straight
away if this compound was as interesting as I thought.
Anyway, in late 2016 Craig’s new PhD student, (now soon-to-be-Dr.) Demetris Bates picked
up Aidan’s work, so I trained him in the use of pyrophorics. He ended up playing the leading synthetic role in this project, as you'll see later.
He is a fantastic chemist, so it took him little time to get to grips with the reaction, optimise its conditions and make a good batch of the compound
above (also in its crystalline form as massive yellow needles).
Demetris gave me a bottle with 20-30 mg of this compound as I thought it could allow the
fabrication of junctions with interesting behaviour. I took the bottle, put it in one of my desk drawers, and completely forgot about it. Yes. I am sorry Demetris,
I completely forgot. In my defence, in 2017 I was an over-caffeinated, sleep-deprived, stressed-out, new-dad PDRA, so some memory impairment was only natural.
More than 2 years later, the stars aligned. In early September 2019 I was finally given an office of my own, so I cleared out my desk – and voila’,
in the back of one of my drawers, I found the little sample bottle Demetris gave me. Chuanli (see here if you don’t have a clue who she is) had just
finished her work on the polyoxometalate molecular wire, and she wanted to have something to do in her last couple of weeks in the UK. Her visit to our
lab was supposed to end on the 30th of September 2019, so I gave her the bottle with Demetris' compound to start a few break-junction experiments. We could see straight
away if this compound was as interesting as I thought.
 Now, why did I think this compound could be interesting? First of all, its conjugation is completely broken. It is like the compound is made by two halves,
only loosely electronically coupled to each other. So it could lead to funny quantum interference effects. Second, it’s a benzil derivative, and benzil is great
fun. It’s a little, simple compound that puzzled physical organic chemists for decades. Even after its structure as C6H5-CO-CO-C6H5 was finally established in
the late 1880s, no one could understand why it had a large-than-expected optical rotatory power, or why it had a moment of dipole of 3.5 Debye in spite of being
symmetrical. IR spectroscopy showed only a single band in the carbonyl area, so everybody agreed the two COs should be identical, and in a trans conformation.
In the end, it turned out that there is a very small splitting in the CO band, so the two carbonyls are not exactly identical. Old spectrophotometers just did
not have enough resolution. This finding, along with the suggestion that the molecule is, in fact, not planar and not in a trans conformation was actually published
in Nature in 1963 (ah, the days when you could get a Nature paper with two IR spectra and 20 lines of text). But some chemists still were not convinced.
The XRD crystal structure was finally published in 1965, and it demonstrated the molecule is neither trans nor cis, but has an intermediate conformation,
associated with large thermal motions. But even that was not enough. in 1987 the gas-phase structure was determined by electron diffraction, and it was shown
that at elevated temperature the structure is almost perfectly trans. Then, a surprising number of theoretical papers was published in the 1980s and 1990s,
analysing the structure of benzil and trying to determine why it had such a strange (and elusive) structure. Solution, solid-state and gas-phase spectroscopic
studies continued to be published, and all together they form today a very nice picture: benzil is an oddball. There is no absolute trans or cis conformation as
there is too much steric hindrance. Instead, there are cisoid (syn) and transoid (anti) conformations, with a very shallow interconversion energy barrier. It looks like it
is very easy for benzil to switch between the two – to the point that it has also been proposed that both cisoid and transoid can coexist in solution (as the higher-energy transoid structure can be stabilised by interactions with the solvent).
So, apart from the chance of adding a piece to a 137-year old scientific puzzle, I thought it would have been extremely interesting to try to force the
syn ⇄ anti interconversion by mechanical means. Mechanically-triggered atropisomerization of a single-molecule junction had already been reported,
and the energies required to achieve such phenomena were greater than the one in place in benzil. Chuanli started by measuring its conductance using the
break-junction technique, that measures the current through a molecular wire as a function of the junction size. It worked better than we thought.
Two clear contributions were evident in the conductance distribution – one corresponding to a smaller junction size of approximately 0.8 nm, and a
second associated with lower conductance, corresponding to longer junctions (approximately 1.2 nm). Some quick-and-dirty MM modelling told us that the
distances were in excellent agreement with the most energetically favoured cisoid conformation (0.8nm) and the ground-state transoid (1.2 nm). So it actually
looked like our model could work.
Chuanli then performed some mechanical modulation experiments, in which a single-molecule junction is repeatedly compressed and
stretched (discussed in more detail here), to show that the process of cisoid ⇄ transoid conversion was reproducible and consistent. It worked.
We could fold our molecular junction like a piece of paper, where the CO-CO bond is our crease. Hence the snappy title of our paper.
This left us with the excellent question: why is the cisoid/syn conformation almost 25 times more conductive than the transoid/anti one?
The game is afoot!
Now, why did I think this compound could be interesting? First of all, its conjugation is completely broken. It is like the compound is made by two halves,
only loosely electronically coupled to each other. So it could lead to funny quantum interference effects. Second, it’s a benzil derivative, and benzil is great
fun. It’s a little, simple compound that puzzled physical organic chemists for decades. Even after its structure as C6H5-CO-CO-C6H5 was finally established in
the late 1880s, no one could understand why it had a large-than-expected optical rotatory power, or why it had a moment of dipole of 3.5 Debye in spite of being
symmetrical. IR spectroscopy showed only a single band in the carbonyl area, so everybody agreed the two COs should be identical, and in a trans conformation.
In the end, it turned out that there is a very small splitting in the CO band, so the two carbonyls are not exactly identical. Old spectrophotometers just did
not have enough resolution. This finding, along with the suggestion that the molecule is, in fact, not planar and not in a trans conformation was actually published
in Nature in 1963 (ah, the days when you could get a Nature paper with two IR spectra and 20 lines of text). But some chemists still were not convinced.
The XRD crystal structure was finally published in 1965, and it demonstrated the molecule is neither trans nor cis, but has an intermediate conformation,
associated with large thermal motions. But even that was not enough. in 1987 the gas-phase structure was determined by electron diffraction, and it was shown
that at elevated temperature the structure is almost perfectly trans. Then, a surprising number of theoretical papers was published in the 1980s and 1990s,
analysing the structure of benzil and trying to determine why it had such a strange (and elusive) structure. Solution, solid-state and gas-phase spectroscopic
studies continued to be published, and all together they form today a very nice picture: benzil is an oddball. There is no absolute trans or cis conformation as
there is too much steric hindrance. Instead, there are cisoid (syn) and transoid (anti) conformations, with a very shallow interconversion energy barrier. It looks like it
is very easy for benzil to switch between the two – to the point that it has also been proposed that both cisoid and transoid can coexist in solution (as the higher-energy transoid structure can be stabilised by interactions with the solvent).
So, apart from the chance of adding a piece to a 137-year old scientific puzzle, I thought it would have been extremely interesting to try to force the
syn ⇄ anti interconversion by mechanical means. Mechanically-triggered atropisomerization of a single-molecule junction had already been reported,
and the energies required to achieve such phenomena were greater than the one in place in benzil. Chuanli started by measuring its conductance using the
break-junction technique, that measures the current through a molecular wire as a function of the junction size. It worked better than we thought.
Two clear contributions were evident in the conductance distribution – one corresponding to a smaller junction size of approximately 0.8 nm, and a
second associated with lower conductance, corresponding to longer junctions (approximately 1.2 nm). Some quick-and-dirty MM modelling told us that the
distances were in excellent agreement with the most energetically favoured cisoid conformation (0.8nm) and the ground-state transoid (1.2 nm). So it actually
looked like our model could work.
Chuanli then performed some mechanical modulation experiments, in which a single-molecule junction is repeatedly compressed and
stretched (discussed in more detail here), to show that the process of cisoid ⇄ transoid conversion was reproducible and consistent. It worked.
We could fold our molecular junction like a piece of paper, where the CO-CO bond is our crease. Hence the snappy title of our paper.
This left us with the excellent question: why is the cisoid/syn conformation almost 25 times more conductive than the transoid/anti one?
The game is afoot!
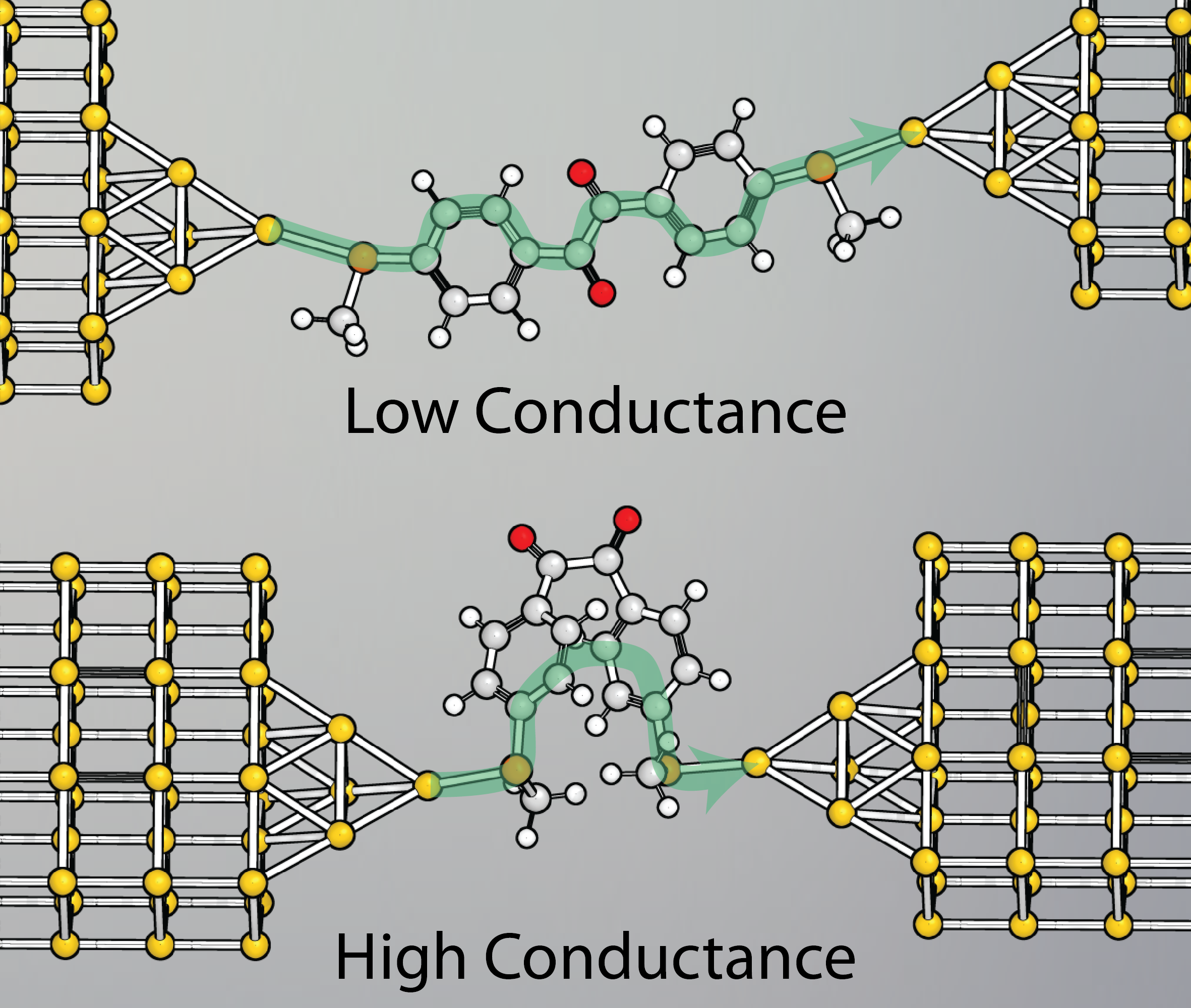 The quick-and-dirty molecular modelling performed earlier showed us that in the cisoid conformation the two phenyl rings also rotate to an almost-cofacial
configuration – similar to what happens in the π-stacking of aromatics. Now, charge transport through stacked phenyl rings has already been demonstrated several times
and a device that would work in a similar way is also discussed in a theoretical paper. So there was a good chance that this optimal
orientation of the two phenyl rings allowed efficient charge transport through their π-π interactions, effectively short-circuiting the molecule. Theoretical calculations based on DFT,
performed by Dr. Hatef Sadeghi and Dr. Sara Sangtarash, also confirmed the model, but we wanted experimental confirmation. That meant trying to measure the
noise power of our junctions, in the two conformations.
Molecular junctions are inherently noisy. We are measuring current flow through something extremely small, and at room temperature. Molecules move and vibrate,
bonds stretch and rock, aromatic rings breathe... everything is in constant motion. And when current goes through all these moving bonds, its magnitude changes
accordingly – in the end, we’re dealing with very noisy signals. But a surprising amount of information is hidden in all that noise. In 2015, an excellent paper was published
in Nano Letters, where the team led by Prof. Latha Venkataraman went on a chase for all this hidden information. They analysed the current signal of molecular
junctions in the domain of frequency to extract numerical information about the “amount” and “colour” of noise. I must admit that as a humble experimental chemist
it took me a good while to understand the manuscript and all the mathematical background, but I guess by now I have a decent grasp of it. What matters here is the
pink noise, the same that is routinely observed in solid-state electronic devices. Pink noise arises from statistical fluctuations and is found almost
everywhere – heartbeat patterns suffer from pink noise, and so does the emission of stars and quasars, neuron firings and even tides and financial systems.
It’s everywhere. But its magnitude is not completely random.
In the most basic terms, what was found is that in a molecular junction where the current follows the molecular bonds, pink noise scales linearly with molecular
conductance. Nothing really surprising here – more current, more noise. But when the current flow takes a “shortcut” and tunnels through space, then the noise
scales with Gn > 1, so not a linear dependence anymore – in this case, highly-conductive junctions are way noisier than expected. So we can extract information about the current
pathways in the molecule by measuring the noise power (a value for the amount of noise) of our nanoelectronic devices and analysing it as a function of molecular conductance.
The quick-and-dirty molecular modelling performed earlier showed us that in the cisoid conformation the two phenyl rings also rotate to an almost-cofacial
configuration – similar to what happens in the π-stacking of aromatics. Now, charge transport through stacked phenyl rings has already been demonstrated several times
and a device that would work in a similar way is also discussed in a theoretical paper. So there was a good chance that this optimal
orientation of the two phenyl rings allowed efficient charge transport through their π-π interactions, effectively short-circuiting the molecule. Theoretical calculations based on DFT,
performed by Dr. Hatef Sadeghi and Dr. Sara Sangtarash, also confirmed the model, but we wanted experimental confirmation. That meant trying to measure the
noise power of our junctions, in the two conformations.
Molecular junctions are inherently noisy. We are measuring current flow through something extremely small, and at room temperature. Molecules move and vibrate,
bonds stretch and rock, aromatic rings breathe... everything is in constant motion. And when current goes through all these moving bonds, its magnitude changes
accordingly – in the end, we’re dealing with very noisy signals. But a surprising amount of information is hidden in all that noise. In 2015, an excellent paper was published
in Nano Letters, where the team led by Prof. Latha Venkataraman went on a chase for all this hidden information. They analysed the current signal of molecular
junctions in the domain of frequency to extract numerical information about the “amount” and “colour” of noise. I must admit that as a humble experimental chemist
it took me a good while to understand the manuscript and all the mathematical background, but I guess by now I have a decent grasp of it. What matters here is the
pink noise, the same that is routinely observed in solid-state electronic devices. Pink noise arises from statistical fluctuations and is found almost
everywhere – heartbeat patterns suffer from pink noise, and so does the emission of stars and quasars, neuron firings and even tides and financial systems.
It’s everywhere. But its magnitude is not completely random.
In the most basic terms, what was found is that in a molecular junction where the current follows the molecular bonds, pink noise scales linearly with molecular
conductance. Nothing really surprising here – more current, more noise. But when the current flow takes a “shortcut” and tunnels through space, then the noise
scales with Gn > 1, so not a linear dependence anymore – in this case, highly-conductive junctions are way noisier than expected. So we can extract information about the current
pathways in the molecule by measuring the noise power (a value for the amount of noise) of our nanoelectronic devices and analysing it as a function of molecular conductance.
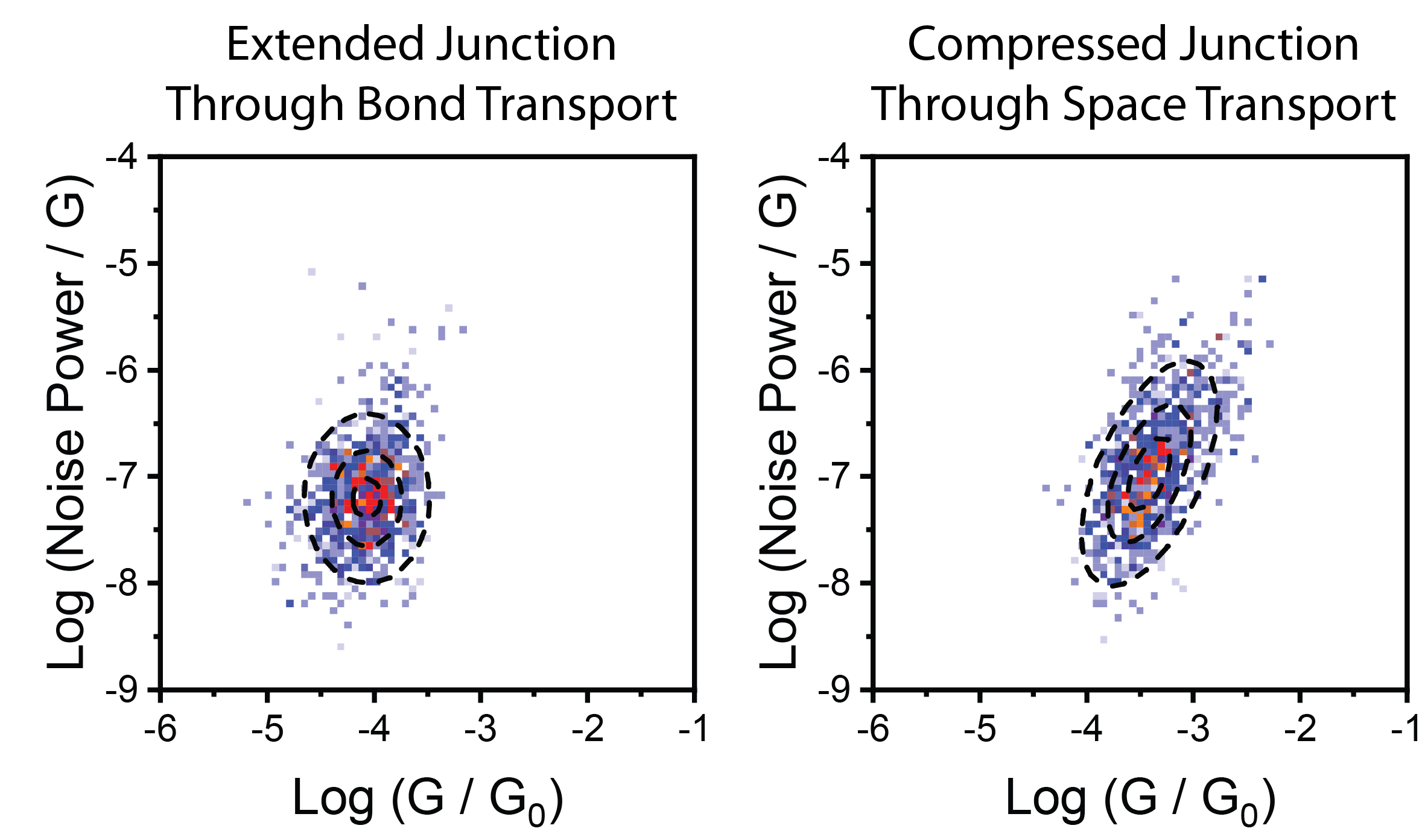 Well, that was no easy feat, and it took us a while to do it well enough. First of all, we had to reduce all other sources of noise.
We already had a decent vibration isolation system, but we improved it a bit to reduce ambient noise by insulating everything with the same
acoustic foam used in recording studios. We ran all electrical devices through a battery-powered uninterrupted power supply, thoroughly checked all common grounds,
and put our STM on top of an active vibration isolation optical table. Before starting the experiment, we thermalised the system to reduce drift by applying continuous
voltage ramps to the piezoelectric transducers for a few hours, to allow them to reach a stable operating temperature. When all of this was done, Chuanli started
recording data. Her experiment was conceptually simple. She would fabricate a junction by creating a 1.25 nm nanogap between the electrodes where the molecule could
only self-assemble in their “extended” transoid (anti) state, and hold it stable for 50 ms. After this time, the piezoelectric transducer would compress the junction by 0.4 nm
and then hold it stable for another 50 ms. If we were correct, this process would result in conversion of the molecule to its cisoid (syn) state. The junction would then be
stretched to rupture to start afresh, and the process repeated while acquiring the current signal at very high speed (we collected 100000 datapoints per second).
As I already discussed here, acquiring data at such speed is not so easy, but this time we were prepared. Instead of storing data as ASCII as we did for our previous experiments,
we directly encoded in binary. The STM workstation is powerful enough to do such encoding on-the-fly, and in our previous experiments it was the
sluggish hard drive write speed that would make our system struggle. And in fact, this time it worked like a charm. Chuanli was able to fabricate tens of thousands of
junctions in relatively little time, finally experiencing (almost) no technical issues! Then we wrote some basic software to do a Fourier Transform on the current signal,
calculate its power spectrum, integrate it to obtain the noise power and, finally, plot its correlation with the junction conductance. The results were absolutely fantastic!
The junction in its extended state showed noise power linearly correlated to the junction conductance. All current
follows the bond pathway. Nothing unexpected. In the compressed state, noise power scaled with G1.6, demonstrating a good amount of through-space charge transport.
And we knew where the through-space bit was – the transport between the two co-facial phenyl rings! We finally had the experimental confirmation we wanted.
We started drafting the paper, and while doing so we also thought of possible control experiments. Demetris synthesised the stilbene analogue, which is completely rigid so
would not show any mechanical modulation of conductance. And it did not. We submitted the paper to a journal and received quite enthusiastic comments,
but we decided to withdraw it as part of the public backlash against the publication of a terrible opinion piece. We then submitted it to Nano Letters, and
this time one of the reviewers suggested to synthesise a compound with only one phenyl ring as further control experiment (see figure below). I wanted to hit myself in the head with a hammer. It was such a good
suggestion I should have thought about it before even writing the paper. But hey, that’s exactly why we do peer review, right?
Well, that was no easy feat, and it took us a while to do it well enough. First of all, we had to reduce all other sources of noise.
We already had a decent vibration isolation system, but we improved it a bit to reduce ambient noise by insulating everything with the same
acoustic foam used in recording studios. We ran all electrical devices through a battery-powered uninterrupted power supply, thoroughly checked all common grounds,
and put our STM on top of an active vibration isolation optical table. Before starting the experiment, we thermalised the system to reduce drift by applying continuous
voltage ramps to the piezoelectric transducers for a few hours, to allow them to reach a stable operating temperature. When all of this was done, Chuanli started
recording data. Her experiment was conceptually simple. She would fabricate a junction by creating a 1.25 nm nanogap between the electrodes where the molecule could
only self-assemble in their “extended” transoid (anti) state, and hold it stable for 50 ms. After this time, the piezoelectric transducer would compress the junction by 0.4 nm
and then hold it stable for another 50 ms. If we were correct, this process would result in conversion of the molecule to its cisoid (syn) state. The junction would then be
stretched to rupture to start afresh, and the process repeated while acquiring the current signal at very high speed (we collected 100000 datapoints per second).
As I already discussed here, acquiring data at such speed is not so easy, but this time we were prepared. Instead of storing data as ASCII as we did for our previous experiments,
we directly encoded in binary. The STM workstation is powerful enough to do such encoding on-the-fly, and in our previous experiments it was the
sluggish hard drive write speed that would make our system struggle. And in fact, this time it worked like a charm. Chuanli was able to fabricate tens of thousands of
junctions in relatively little time, finally experiencing (almost) no technical issues! Then we wrote some basic software to do a Fourier Transform on the current signal,
calculate its power spectrum, integrate it to obtain the noise power and, finally, plot its correlation with the junction conductance. The results were absolutely fantastic!
The junction in its extended state showed noise power linearly correlated to the junction conductance. All current
follows the bond pathway. Nothing unexpected. In the compressed state, noise power scaled with G1.6, demonstrating a good amount of through-space charge transport.
And we knew where the through-space bit was – the transport between the two co-facial phenyl rings! We finally had the experimental confirmation we wanted.
We started drafting the paper, and while doing so we also thought of possible control experiments. Demetris synthesised the stilbene analogue, which is completely rigid so
would not show any mechanical modulation of conductance. And it did not. We submitted the paper to a journal and received quite enthusiastic comments,
but we decided to withdraw it as part of the public backlash against the publication of a terrible opinion piece. We then submitted it to Nano Letters, and
this time one of the reviewers suggested to synthesise a compound with only one phenyl ring as further control experiment (see figure below). I wanted to hit myself in the head with a hammer. It was such a good
suggestion I should have thought about it before even writing the paper. But hey, that’s exactly why we do peer review, right?
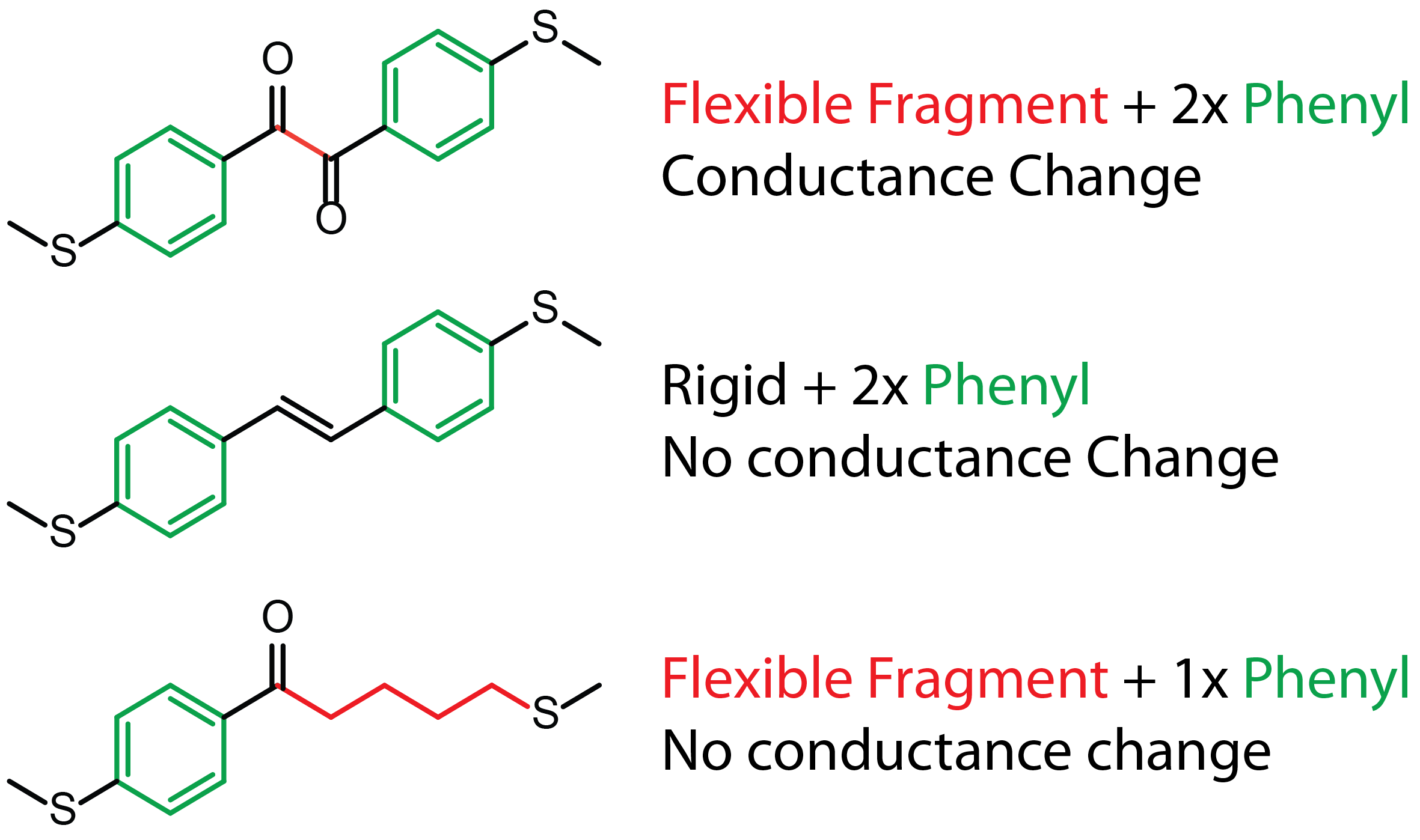 In the compound suggested by the reviewer there would be no π-π interaction upon junction compression,
so conductance should not increase significantly and there should not be increased through-space charge transport. Demetris the synthetic wizard went once again back to the
bench and made the compound in very little time (well, once he had the necessary starting materials!).
I measured its conductance, electromechanical properties, and power spectral density - everything checked out.
Very little conductance increase upon compression, and noise analysis showing no through-bond to through-space charge transport transition. And the rest is just
boring office work: edit the paper, prepare new figures, add loads of stuff to the SI, write a rebuttal letter, etc… and the paper was accepted. A quick thank you
to the editorial staff at Nano Letters – they granted us a revision extension when we had issues in sourcing the starting materials for the synthesis of the control
molecule due to the ongoing coronavirus pandemic (I understand it’s hard times for us all, but 4 weeks to deliver us a simple compound marked “in stock” on the supplier
website is a bit too long!!!), and the revision/peer-review/proof turnover was lightning-fast.
In the end, this project turned out to be another monstruously-sized one. Chuanli and I collected and analysed more than 93 GB of data, and this time all of it is presented in the paper,
in a way or another. As Sir Arthur Conan Doyle made his most famous character once say:
“DATA! DATA! DATA! I can’t make bricks without clay!”
In the compound suggested by the reviewer there would be no π-π interaction upon junction compression,
so conductance should not increase significantly and there should not be increased through-space charge transport. Demetris the synthetic wizard went once again back to the
bench and made the compound in very little time (well, once he had the necessary starting materials!).
I measured its conductance, electromechanical properties, and power spectral density - everything checked out.
Very little conductance increase upon compression, and noise analysis showing no through-bond to through-space charge transport transition. And the rest is just
boring office work: edit the paper, prepare new figures, add loads of stuff to the SI, write a rebuttal letter, etc… and the paper was accepted. A quick thank you
to the editorial staff at Nano Letters – they granted us a revision extension when we had issues in sourcing the starting materials for the synthesis of the control
molecule due to the ongoing coronavirus pandemic (I understand it’s hard times for us all, but 4 weeks to deliver us a simple compound marked “in stock” on the supplier
website is a bit too long!!!), and the revision/peer-review/proof turnover was lightning-fast.
In the end, this project turned out to be another monstruously-sized one. Chuanli and I collected and analysed more than 93 GB of data, and this time all of it is presented in the paper,
in a way or another. As Sir Arthur Conan Doyle made his most famous character once say:
“DATA! DATA! DATA! I can’t make bricks without clay!”
Many thanks to soon-to-be-Dr. Demetris Bates for his synthetic wizardry, to Dr. Chuanli Wu for her relentless torture of our STM, and to Prof. Simon J. Higgins for proofreading my Italenglish.
16/04/2020
A Chemically Soldered Polyoxometalate Single-Molecule Transistor
And here I am again, with another paper just published in Angewandte Chemie. This work hasn’t been an emotional rollercoaster like the
“hemilabile ligand for molecular electronics” that I described in August last year, but it has been an absolute mammoth of data
acquisition and analysis, performed with incredible perseverance and diligence by soon-to-be-Dr. Chuanli Wu.
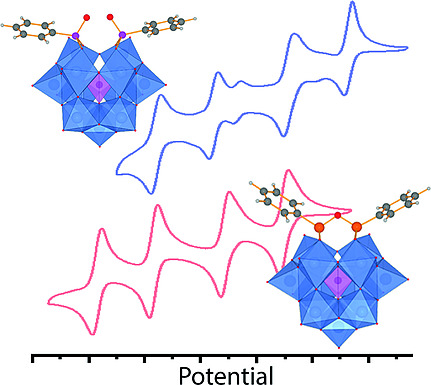 It all started with a small pot of money I was given in 2018 for the synthesis of some cluster-containing molecular wires.
The idea was to use these funds to gain some skills in small, atomically defined cluster preparation and purification
(somewhat different from the chemistry I did up to then), and to use these to collect single-cluster conductance data
that could underpin my fellowship applications. One of the reviewers for my Future Leaders Fellowship application (alas, unsuccessful) raised doubts on
the stability of clusters under an intense electrical field, such as the one that is experienced by a molecular junction,
and I wanted to show that these can be extremely resilient species. After the funds were released, I went back to the bench
and prepared a few different classes of clusters – from pure metallic materials containing copper, silver or ruthenium,
to metal chalcogenides and metal oxides. The latter, perhaps better known as polyoxometalates, interested me for their
electrochemical behaviour – some of the larger ones have multiple, well-defined oxidation states, and their cyclic
voltammograms (CV) can be oddly fascinating (see here on the right for an example from a Keggin polyoxoanion) as
all the different charge states give very well-defined reversible redox processes and therefore many pleasant waves in the CV!
It all started with a small pot of money I was given in 2018 for the synthesis of some cluster-containing molecular wires.
The idea was to use these funds to gain some skills in small, atomically defined cluster preparation and purification
(somewhat different from the chemistry I did up to then), and to use these to collect single-cluster conductance data
that could underpin my fellowship applications. One of the reviewers for my Future Leaders Fellowship application (alas, unsuccessful) raised doubts on
the stability of clusters under an intense electrical field, such as the one that is experienced by a molecular junction,
and I wanted to show that these can be extremely resilient species. After the funds were released, I went back to the bench
and prepared a few different classes of clusters – from pure metallic materials containing copper, silver or ruthenium,
to metal chalcogenides and metal oxides. The latter, perhaps better known as polyoxometalates, interested me for their
electrochemical behaviour – some of the larger ones have multiple, well-defined oxidation states, and their cyclic
voltammograms (CV) can be oddly fascinating (see here on the right for an example from a Keggin polyoxoanion) as
all the different charge states give very well-defined reversible redox processes and therefore many pleasant waves in the CV!
 For a cluster to be used efficiently in molecular junctions it needs to be terminated with aurophilic groups, which can form
coordination bonds with two nanoelectrodes and close the circuit. This somewhat limits the range of polyoxometalates I could
use, as their functionalisation with organic ligands is not exactly straightforward. I started by synthesising a Lindqvist
polyoxovanadate (see XRD structure on the left) and I measured its conductance – it proved to be quite poorly conductive, and a bit too close
to the noise level of our instruments at the time to be studied in more detail, but it gave me the initial data I needed for
my fellowship applications, which I’m sure helped me greatly in securing the Royal Society URF position.
That it was so insulating wasn’t surprising – most bulk metal oxides are good insulators, and the Lindqvist anion proved
to be no exception. By then, however, I really wanted to find a redox-active cluster that could study at the single-molecule
level in an electrochemical environment. A literature search turned up a small polyoxomolybdate, having Anderson-Evans structure,
that could be functionalised in an optimal way, with the two organic ligands coordinated to opposite sides of a MnO6 octahedron.
The synthesis looked straightforward on paper, but it took me a few weeks before I had a sample pure enough for characterisation
and crystal growing – which was good: all useful skills I had to learn if I wanted to work with more of these cluster materials.
For a cluster to be used efficiently in molecular junctions it needs to be terminated with aurophilic groups, which can form
coordination bonds with two nanoelectrodes and close the circuit. This somewhat limits the range of polyoxometalates I could
use, as their functionalisation with organic ligands is not exactly straightforward. I started by synthesising a Lindqvist
polyoxovanadate (see XRD structure on the left) and I measured its conductance – it proved to be quite poorly conductive, and a bit too close
to the noise level of our instruments at the time to be studied in more detail, but it gave me the initial data I needed for
my fellowship applications, which I’m sure helped me greatly in securing the Royal Society URF position.
That it was so insulating wasn’t surprising – most bulk metal oxides are good insulators, and the Lindqvist anion proved
to be no exception. By then, however, I really wanted to find a redox-active cluster that could study at the single-molecule
level in an electrochemical environment. A literature search turned up a small polyoxomolybdate, having Anderson-Evans structure,
that could be functionalised in an optimal way, with the two organic ligands coordinated to opposite sides of a MnO6 octahedron.
The synthesis looked straightforward on paper, but it took me a few weeks before I had a sample pure enough for characterisation
and crystal growing – which was good: all useful skills I had to learn if I wanted to work with more of these cluster materials.
 Just as I was finishing preparing these compounds, Chuanli, who was visiting our laboratory at the time, had also finished collecting
all the data she needed for a project she was working on – the study of the in-situ formation of hydrogen-bonding chains during
break-junction experiments (published a few months ago in Nanoscale). Before this work, she had worked quite extensively on the
in-situ electrochemistry of NiO, in a project that was very challenging but, alas, resulted only in more questions than answers.
Nickel is indeed a weird metal. While partially unsuccessful, she gained a lot of experience in the use of the electrochemical
scanning tunnelling microscope, and she decided to take on the challenge of measuring the polyoxometalate single-molecule
conductance under electrochemical potential control. The idea here was to probe the charge transport properties of the
polyoxometalate in its various electrochemical states. Initial cyclic voltammetry showed us we could both oxidise and reduce
the Anderson-Evans cluster from its natural -3 state, to either -2 or -4. And, as usual, our problems started. The polyoxomolybdate
wasn’t that soluble in the solvents we regularly use in molecular electronics studies, and the necessity of an ionic environment
(an electrolyte) further complicated the issue. Chuanli started screening a few solvents where we knew we could get an electrochemical
response – acetonitrile, propylene carbonate, DMF, etc. – but each of them presented an issue in the single-molecule measurements
we wanted to perform. They either resulted in too much background noise, dissolved the electrodes coating, prevented formation of
junctions or gave an unreliable electrochemical potential (a problem in most non-aqueous electrochemical studies).
Just as I was finishing preparing these compounds, Chuanli, who was visiting our laboratory at the time, had also finished collecting
all the data she needed for a project she was working on – the study of the in-situ formation of hydrogen-bonding chains during
break-junction experiments (published a few months ago in Nanoscale). Before this work, she had worked quite extensively on the
in-situ electrochemistry of NiO, in a project that was very challenging but, alas, resulted only in more questions than answers.
Nickel is indeed a weird metal. While partially unsuccessful, she gained a lot of experience in the use of the electrochemical
scanning tunnelling microscope, and she decided to take on the challenge of measuring the polyoxometalate single-molecule
conductance under electrochemical potential control. The idea here was to probe the charge transport properties of the
polyoxometalate in its various electrochemical states. Initial cyclic voltammetry showed us we could both oxidise and reduce
the Anderson-Evans cluster from its natural -3 state, to either -2 or -4. And, as usual, our problems started. The polyoxomolybdate
wasn’t that soluble in the solvents we regularly use in molecular electronics studies, and the necessity of an ionic environment
(an electrolyte) further complicated the issue. Chuanli started screening a few solvents where we knew we could get an electrochemical
response – acetonitrile, propylene carbonate, DMF, etc. – but each of them presented an issue in the single-molecule measurements
we wanted to perform. They either resulted in too much background noise, dissolved the electrodes coating, prevented formation of
junctions or gave an unreliable electrochemical potential (a problem in most non-aqueous electrochemical studies).
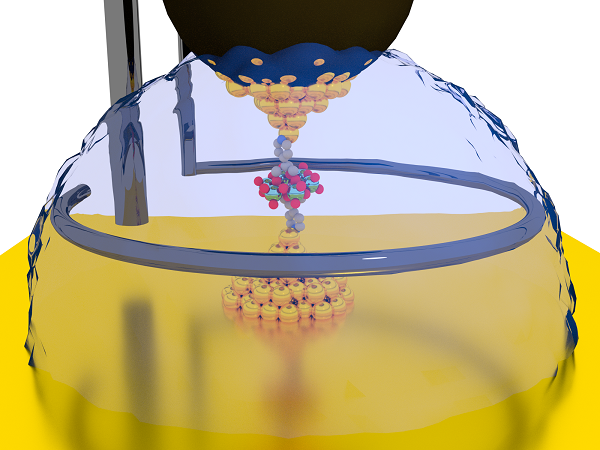 At that time, Xiaohang Qiao (a PhD student working with us) was studying the use of deep eutectic mixtures in molecular electronics.
These are excellent solvents, they provide an ionic environment ideally suited to electrochemistry, and showed good promise for
STM work. So Chuanli and Xiaohang teamed up and started studying the polyoxometalate in a deep eutectic mixture of choline
chloride and urea. This started to give us quite good results: the polyoxomolybdate was soluble, we could form junctions in
the electrochemical environment (made of 4 electrodes, two Au as source/drain and two Pt as counter/reference as in the figure
here), and we could see a difference in the conductance as the potential was made more negative and the polyoxometalate charge
state was changed from -3 to -4. Furthermore, the presence of many chloride ions allowed us to use a chloridised silver wire as
reference electrode, with excellent stability. Unfortunately, they then had issues at positive potentials: the excess chloride
ions that gave us the benefit of a stable reference electrode also caused the Au electrodes to oxidise and dissolve before we
could access the -2 state in this setup.
At that time, Xiaohang Qiao (a PhD student working with us) was studying the use of deep eutectic mixtures in molecular electronics.
These are excellent solvents, they provide an ionic environment ideally suited to electrochemistry, and showed good promise for
STM work. So Chuanli and Xiaohang teamed up and started studying the polyoxometalate in a deep eutectic mixture of choline
chloride and urea. This started to give us quite good results: the polyoxomolybdate was soluble, we could form junctions in
the electrochemical environment (made of 4 electrodes, two Au as source/drain and two Pt as counter/reference as in the figure
here), and we could see a difference in the conductance as the potential was made more negative and the polyoxometalate charge
state was changed from -3 to -4. Furthermore, the presence of many chloride ions allowed us to use a chloridised silver wire as
reference electrode, with excellent stability. Unfortunately, they then had issues at positive potentials: the excess chloride
ions that gave us the benefit of a stable reference electrode also caused the Au electrodes to oxidise and dissolve before we
could access the -2 state in this setup.
We then remembered that a few years ago our lab published a
series
of papers
in JACS
(I helped out on the third in the series)
on the use of ionic liquids in single-molecule electronics. They had everything we were looking for: powerful solvents with a
wide electrochemical window and a simple Pt wire can be used as reference electrode with excellent stability – thinking back,
I don’t really know why we didn’t use these from the beginning.
Ah yes, ionic liquids are quite a nightmare to purify. And expensive. Very expensive.
Ionic liquids tend to pick up water quite quickly – when they’re wet, the electrochemical window narrows down a lot, and we
needed all of it, or at least 2.5 V. Also, last year we weren’t exactly swimming in cash. Luckily, we had about 50-75 mL
of 1-butyl-3-methylimidazolium trifluoromethanesulfonate in our glove box, leftover from our previous studies – while it sat
there for more than 4 years, it couldn’t have gone too bad in an inert, dry atmosphere. We cleaned it up, testing its
electrochemistry until it was pure (and it took some time!), and then Chuanli started collecting data. This setup worked like a charm!
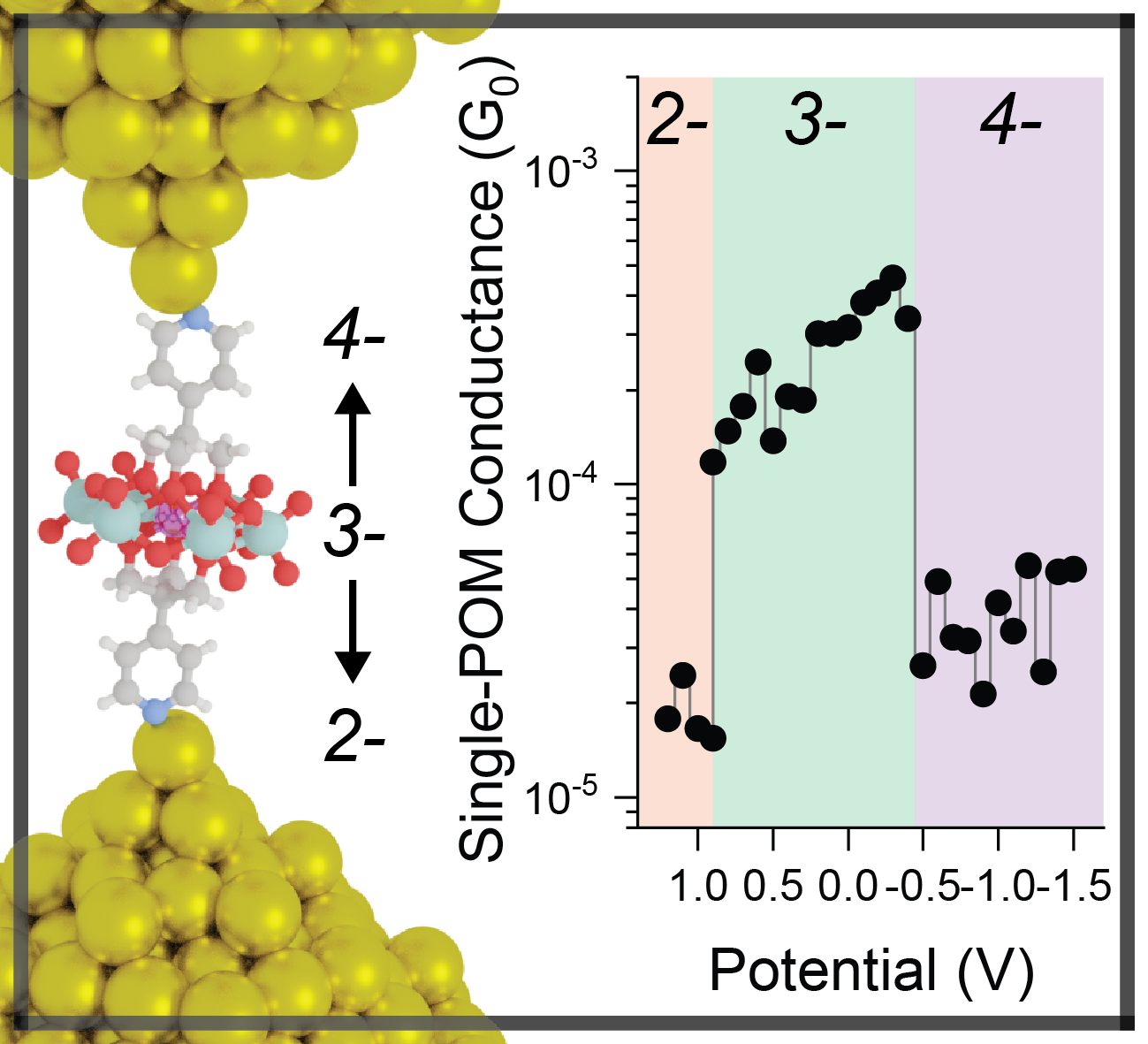 As you can read in the paper, using an ionic liquid allowed us to probe conductance over 2.7 V of electrochemical potential,
where we could measure the polyoxometalate in all three states (-2, -3 and -4). We found a rather unusual response – as the
cluster was oxidised or reduced from its native -3 state, conductance suddenly dropped by one order of magnitude.
From our data, it looked like the mechanism of charge transport was phase-coherent tunnelling, and the polyoxometalate
in different charge states presented a different barrier to charge tunnelling. While not a novel phenomenon (see examples
here,
here and
here),
it was pretty exciting to find out such a behaviour
in a polyoxometalate. To further strengthen our claim, we did some modelling, which required quite a lot of data analysis
(good thing I had an M.2 NVMe solid state disk with a write speed of ~1.9 GB/s or this would have taken years), and some
more experiments, which you can read about in the paper (that is open access and free to read to everyone).
As you can read in the paper, using an ionic liquid allowed us to probe conductance over 2.7 V of electrochemical potential,
where we could measure the polyoxometalate in all three states (-2, -3 and -4). We found a rather unusual response – as the
cluster was oxidised or reduced from its native -3 state, conductance suddenly dropped by one order of magnitude.
From our data, it looked like the mechanism of charge transport was phase-coherent tunnelling, and the polyoxometalate
in different charge states presented a different barrier to charge tunnelling. While not a novel phenomenon (see examples
here,
here and
here),
it was pretty exciting to find out such a behaviour
in a polyoxometalate. To further strengthen our claim, we did some modelling, which required quite a lot of data analysis
(good thing I had an M.2 NVMe solid state disk with a write speed of ~1.9 GB/s or this would have taken years), and some
more experiments, which you can read about in the paper (that is open access and free to read to everyone).
What I think is staggering about this project is the sheer amount of work performed by Chuanli. In the paper, we present
data from approximately 100000 (yes, one hundred thousand!) single-molecule devices she fabricated under electrochemical
control, and an additional 20000 devices fabricated under source-drain bias modulation. But this is just the tip of the
iceberg. There are many “preliminary measurements” in other solvents (the ones that didn’t really worked as we wanted),
and from the sheer size of her “POM” folder (sitting on my back-up disk at 11.3 GB zipped at ~20% rate) I estimate at least
another 200000 junctions were fabricated for this project. And I’m being conservative!
Anyway, the publishing process was refreshingly quick and straightforward - so this story ends in a quite anticlimactic fashion.
We had an initial desk rejection from another journal, but after submitting to Angewandte Chemie we received very good feedback from the reviewers –
they asked for a single control experiment which we hadn’t done. We did it, it worked as expected, and we finally got this beast published.
I hope you’ll enjoy reading about our work as much as I enjoyed writing about it – honestly, with experimental data as good as the one Chuanli collected,
the paper almost wrote itself.
Many thanks to soon-to-be-Dr. Chuanli Wu for for keeping up with my continuous “MORE DATA!” requests for many months, and to Prof. Richard J. Nichols and Prof. Simon J. Higgins for their help in putting this together. Edited 26/05/2020 to correct a few typos. You never proofread enough! Re-edited 06/02/2026 because today I learned the correct spelling is "anticlimactic"...
01/08/2019
Hemilabile Ligands as Mechanosensitive Electrode Contacts for Molecular Electronics
So, I'll start this column by writing about this paper here, that we recently published in Angewandte Chemie.
As you'll see, this is a nice story with failures, puzzlement and despair, but also great teamwork and sustained effort. It follows a quite oftenly encountered scientific path: we started
looking into some compounds to test one particular theory (quantum interference) but instead, we found something
completely different (mechanoresistance) but equally exciting, that can also help
us to understand some earlier puzzling results we (and other research groups around the world) had obtained. In some ways, it is also a manifestation of
Westheimer's Discovery
"Why spend a day in the library when you can learn the same thing by working in the laboratory for a month?".
 The bulk of this work was performed whilst I was working as a PDRA for Prof. Richard Nichols,
and I was training one of Prof. Simon J. Higgins (my PhD supervisor) students in the group, now Dr. Nicolo' Ferri.
He picked up a project I left incomplete at the end of my PhD (late 2014-early 2015) as I hadn't been able to synthesise
one of the key compounds, a fused bithiophene with a bridging carbonyl function and methyl thioether contact groups for binding to gold electrodes.
The target was quite interesting because it had a very narrow bandgap and theory had predicted some odd quantum interference effects from the carbonyl,
that would result in unusually high (or unusually low, depending on which theoretician we listened to...) conductance.
Following a few recipes for apparently similar compounds I found in the literature just gave a black sticky mess,
and after a few disappointing attempts, I started my PDRA working on molecule/semiconductor interfaces and I completely forgot about the whole project.
Just to explain what kind of chemist Nicolo' is, he not only willingly accepted this work that gave me only pain and disappointment,
but actually managed to synthesise this key compound, grew beautiful (and big) shiny purple crystals, got the SCXRD that confirmed the structure,
did some more synthetic work, and performed all the measurements that you can find in the above paper.
The bulk of this work was performed whilst I was working as a PDRA for Prof. Richard Nichols,
and I was training one of Prof. Simon J. Higgins (my PhD supervisor) students in the group, now Dr. Nicolo' Ferri.
He picked up a project I left incomplete at the end of my PhD (late 2014-early 2015) as I hadn't been able to synthesise
one of the key compounds, a fused bithiophene with a bridging carbonyl function and methyl thioether contact groups for binding to gold electrodes.
The target was quite interesting because it had a very narrow bandgap and theory had predicted some odd quantum interference effects from the carbonyl,
that would result in unusually high (or unusually low, depending on which theoretician we listened to...) conductance.
Following a few recipes for apparently similar compounds I found in the literature just gave a black sticky mess,
and after a few disappointing attempts, I started my PDRA working on molecule/semiconductor interfaces and I completely forgot about the whole project.
Just to explain what kind of chemist Nicolo' is, he not only willingly accepted this work that gave me only pain and disappointment,
but actually managed to synthesise this key compound, grew beautiful (and big) shiny purple crystals, got the SCXRD that confirmed the structure,
did some more synthetic work, and performed all the measurements that you can find in the above paper.
Now, for the organikers out there, we initially tried to synthesise this in a quite unpleasant sequence, where all steps involved either something toxic,
something smelly or something pyrophoric (well, it's just n-BuLi, but still...)! Starting from cheap-ish 2-bromothiophene he had to do:
- Old-school Ni-catalysed Kumada coupling to get 2,2'-bithiophene
- Tetrabromination of 2,2'-bithiophene with elemental Br to give 3,3',5,5'-tetrabromo-2,2'-bithiophene
- Double selective halogen-lithium exchange and quench with dimethyl disulfide (smell!!!) to give 3,3'-dibromo-5,5'-bis(methylthio)-2,2'-bithiophene
- Double lithium-halogen exchange with n-BuLi and reaction with an appropriate carbonyl source
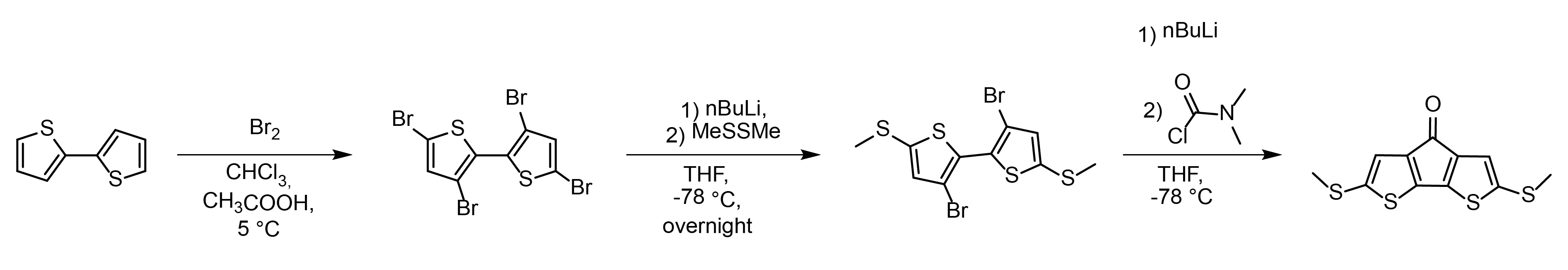
So, when Nicolo' tackled this, I had already tried a few carbonyl sources, and I was so desperate I even tried using phosgene at some point(!). Just as a control experiment, Nicolo' tried the published route to the parent compound lacking the two thiomethyl moieties, and it worked perfectly - it was evident that the two -SMe contact groups were messing up the electronic structure of one of the reaction intermediates and prevented efficient ring-closure to a cyclopentadienone. Obviously, it proved impossible to install the two -SMe on the already-made cyclopentadithiophen-4-one - bromination followed by C-S couplings, lithiations, direct -SMe attack using the infamous NaSMe, etc. all failed. This is enough to drive someone mad! Nicolo' decided to go back to the original route we designed (but with commercial 2,2'-bithiophene from Fluorochem as it was cheap enough), and try again the double lithiation of 3,3',5,5'-tetrabromo-2,2'-bithiophene followed by treatment with dimethylcarbamoyl chloride and quench with ammonium chloride. He carefully prepared all the starting materials, and I remember him recrystallising 25g of 3,3',5,5'-tetrabromo-2,2'-bithiophene in a massive round bottom flask I didn't even know was still knocking about in the lab. After a few horrendously unsuccessful attempts, where he varied the reaction conditions to better understand what was going on, he found an obscure Japanese paper (apologies, I can't find the reference now!) discussing other similar ring-closure reactions to cyclopentadienones. In some of the preparations discussed in the paper, the final quench with ammonium chloride (designed to kick out the -N(Me)2 group and give the cyclopentadienone ring) was performed, with no justifying comment or discussion, at -40° C instead of the room temperature reported in all the other recipes we were following. When he tried this, the reaction turned immediately a lovely shade of pink, indicating a low-bandgap material was actually present in the flask, and at the end, after a chromatographic column and careful 2-layers crystal growing, he obtained lovely deep-purple block crystals in 80% yield.
So, all excited about this compound, we did some measurements on its single-molecule charge transport properties. They were an absolute mess. The compound gave conductance values spanning more than two orders of magnitude, making any kind of statistical analysis to determine the most probable conductance quite meaningless. Nicolo' was quite gutted - he spent so much time getting here, making those lovely purple crystals, and now everything turned sour. The results were absolutely horrible.
 We started to scour the literature looking for an explanation,
and we found out we weren't the first noticing a peculiar behaviour in
compounds incorporating terminal thiophenes. Unusually wide conductance histograms were already reported here,
and odd effects were found in another paper. However, we weren't completely happy with the
explanations given - Simon also pointed out that the thiophene unit could potentially act as additional contact to the electrodes, as it is
known it can coordinate, albeit weakly, to transition metals, and reports on the use of thiophenes themselves as contact groups for molecular junctions are aplenty.
So the idea we came up with was that the unusually large conductance span was due to an abundance of anchoring places for the electrodes,
and therefore the single-molecule junctions weren't as defined as we originally hoped. To test this idea, however,
we had to do some serious work on our instruments, to give them more versatility and perform some modulation experiments,
where we would vibrate the electrodes to compress and relax the junction with a precision of a few Angstroms, thereby forcing the molecule to adapt different
conformations at the electrode interface. Luckily, in late 2016, I visited Prof. Bingqian Xu's laboratory, where they were doing this kind of experiments
as part of an ongoing collaboration, and I had more-or-less understood what I had to do to improve our instruments.
Armed with an arbitrary waveform generator,
a data acquisition board,
a wide-bandwidth preamplifier,
a dozen BNC cables and a very shallow understanding of how Labview works, I opened our STM, fitted new connections, designed a couple of PCBs
(so many thanks to Matty Henderson in our electronic workshop!), got them developed, painstakingly soldered some tiny
Hirose U-FL connectors (I absolutely hate them!), wired everything, wrote a (very!) crude Labview VI and BAM!
We had the instrument we needed. We could drive our system's electrodes with modulations in positions at frequencies up to 10 kHz
(higher than that, we would smash our piezo transducers) with sub-Angstrom precision. We tested it for a few weeks, to ensure
we weren't damaging the piezos and to check for data consistency with the literature and, when we were finally happy,
we did the experiments we wanted. And they worked!
We started to scour the literature looking for an explanation,
and we found out we weren't the first noticing a peculiar behaviour in
compounds incorporating terminal thiophenes. Unusually wide conductance histograms were already reported here,
and odd effects were found in another paper. However, we weren't completely happy with the
explanations given - Simon also pointed out that the thiophene unit could potentially act as additional contact to the electrodes, as it is
known it can coordinate, albeit weakly, to transition metals, and reports on the use of thiophenes themselves as contact groups for molecular junctions are aplenty.
So the idea we came up with was that the unusually large conductance span was due to an abundance of anchoring places for the electrodes,
and therefore the single-molecule junctions weren't as defined as we originally hoped. To test this idea, however,
we had to do some serious work on our instruments, to give them more versatility and perform some modulation experiments,
where we would vibrate the electrodes to compress and relax the junction with a precision of a few Angstroms, thereby forcing the molecule to adapt different
conformations at the electrode interface. Luckily, in late 2016, I visited Prof. Bingqian Xu's laboratory, where they were doing this kind of experiments
as part of an ongoing collaboration, and I had more-or-less understood what I had to do to improve our instruments.
Armed with an arbitrary waveform generator,
a data acquisition board,
a wide-bandwidth preamplifier,
a dozen BNC cables and a very shallow understanding of how Labview works, I opened our STM, fitted new connections, designed a couple of PCBs
(so many thanks to Matty Henderson in our electronic workshop!), got them developed, painstakingly soldered some tiny
Hirose U-FL connectors (I absolutely hate them!), wired everything, wrote a (very!) crude Labview VI and BAM!
We had the instrument we needed. We could drive our system's electrodes with modulations in positions at frequencies up to 10 kHz
(higher than that, we would smash our piezo transducers) with sub-Angstrom precision. We tested it for a few weeks, to ensure
we weren't damaging the piezos and to check for data consistency with the literature and, when we were finally happy,
we did the experiments we wanted. And they worked! In our experiments, we modulated the position of the electrodes by just 3 Angstroms,
and we observed large changes in conductance as the junction was compressed (high G state) or relaxed (low G state).
So our idea was almost proven: as the thiophene was brought in proximity to the metallic electrode it interacted with the electrode and increased the electronic coupling,
acting like the labile part of a hemilabile ligand.
As the junction was relaxed, the electrode-thiophene distance increased and any interaction died out, giving a lower coupling and lower conductance.
Nicolo' synthesised a few more compounds, either for control experiments (a biphenylene that didn't show much change in conductance during modulation experiments),
or to demonstrate the versatility of our chemical design and to show some degree of chemical control (i.e. he replaced the carbonyl with a
gem-dimethylmethylene group which gave even higher conductance modulations), and the brilliant undergraduate student
we had in the lab (Maeve McLaughlin) at the time gave a hand with the synthesis. We improved the measurement methods, wrote better software, and kept
improving our hardware, as we also wanted to do some very high-frequency modulations to push our instrument to its limits. That wasn't so easy to do
as we started to have problems with the data acquisition - it turns out acquiring 24-bit data from 4 channels at 215000 samples per second is quite demanding!
However, after some software optimisation, in which I learned A LOT about Labview (and started following DataFlow G on twitter),
we were able to obtain the data we presented in the paper, where we drove our junctions at 10 kHz modulations of 3 Angstrom for over 100 ms. Which was pretty cool.
In our experiments, we modulated the position of the electrodes by just 3 Angstroms,
and we observed large changes in conductance as the junction was compressed (high G state) or relaxed (low G state).
So our idea was almost proven: as the thiophene was brought in proximity to the metallic electrode it interacted with the electrode and increased the electronic coupling,
acting like the labile part of a hemilabile ligand.
As the junction was relaxed, the electrode-thiophene distance increased and any interaction died out, giving a lower coupling and lower conductance.
Nicolo' synthesised a few more compounds, either for control experiments (a biphenylene that didn't show much change in conductance during modulation experiments),
or to demonstrate the versatility of our chemical design and to show some degree of chemical control (i.e. he replaced the carbonyl with a
gem-dimethylmethylene group which gave even higher conductance modulations), and the brilliant undergraduate student
we had in the lab (Maeve McLaughlin) at the time gave a hand with the synthesis. We improved the measurement methods, wrote better software, and kept
improving our hardware, as we also wanted to do some very high-frequency modulations to push our instrument to its limits. That wasn't so easy to do
as we started to have problems with the data acquisition - it turns out acquiring 24-bit data from 4 channels at 215000 samples per second is quite demanding!
However, after some software optimisation, in which I learned A LOT about Labview (and started following DataFlow G on twitter),
we were able to obtain the data we presented in the paper, where we drove our junctions at 10 kHz modulations of 3 Angstrom for over 100 ms. Which was pretty cool.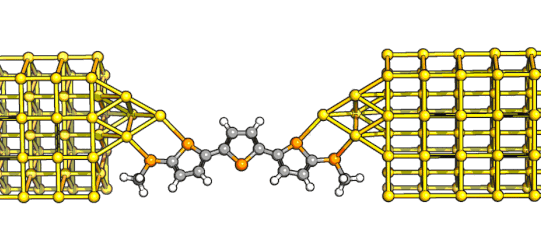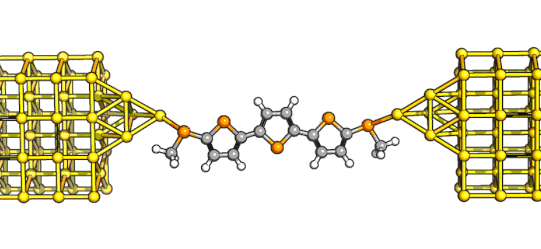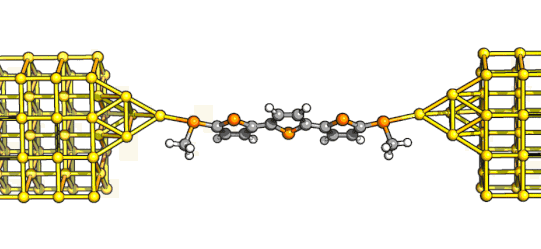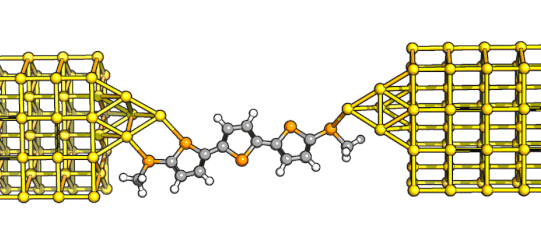 After all this experimental work, we needed a confirmation from theory to show that it was these
thiophene-metal interactions that were indeed responsible for the observed behaviour. Our close collaborators in Lancaster,
Prof. Colin J. Lambert,
Dr. Sara Sangtarash and
Dr. Hatef Sadeghi developed the model that explained the observed phenomena.
They found that when the junctions with thiomethylthiophene terminal groups are compressed
there are indeed increased interactions, as can be seen in the figure here. The interactions result in a stronger contact, more electrically transparent, that leads
to a higher conductance. So, FINALLY, everything made sense. Time to write it up as a paper and start the neverending roulette of choosing a journal → formatting
the manuscript → submit → peer-review → revise → resubmit → outcome = Y/N? (Y = celebrate; N = back to choosing a journal).
The paper was actually rejected on first submission, but it eventually found a very cosy home at Angewandte Chemie.
After all this experimental work, we needed a confirmation from theory to show that it was these
thiophene-metal interactions that were indeed responsible for the observed behaviour. Our close collaborators in Lancaster,
Prof. Colin J. Lambert,
Dr. Sara Sangtarash and
Dr. Hatef Sadeghi developed the model that explained the observed phenomena.
They found that when the junctions with thiomethylthiophene terminal groups are compressed
there are indeed increased interactions, as can be seen in the figure here. The interactions result in a stronger contact, more electrically transparent, that leads
to a higher conductance. So, FINALLY, everything made sense. Time to write it up as a paper and start the neverending roulette of choosing a journal → formatting
the manuscript → submit → peer-review → revise → resubmit → outcome = Y/N? (Y = celebrate; N = back to choosing a journal).
The paper was actually rejected on first submission, but it eventually found a very cosy home at Angewandte Chemie.In brief, and for the fans of tl;dr - we made a hellish small molecule, it didn't do what we wanted it to do, so we tortured it until it behaved and we published the results here.
Many thanks to Dr. Nicolo' Ferri, Prof. Richard J. Nichols and Prof. Simon J. Higgins for their help in putting this together, and for their thorough proofread.