The free-electron-like state observed in a scanning tunnelling spectroscopy study of a chiral p(2×4) monolayer of glycinate ions on the copper (100) surface [Kanazawa K et al 2007 J. Am. Chem. Soc. 129 740] is shown from density functional theory calculations to originate from a copper Shockley surface state at the surface Brillouin zone boundary of the clean surface with highly anisotropic dispersion. The presence of the glycinate ions on the surface causes a dramatically enhanced tunnelling into this surface state that is otherwise not observed in tunnelling on the bare surface.
Advances in molecular assembly experiments on metal surfaces [1] and potential applications arising from them [2] call for a better understanding of the electronic structure at the interface of metals and organic systems. In particular, interest in delocalized electronic states is high, because of their potential use in molecular and opto-electronics applications. Recently, free-electron-like states arising in layers of organic molecules on metal surfaces at energies close to the Fermi energy have been observed by scanning tunnelling and photoemission spectroscopies [3,4,5]. In systems where the molecule is more or less physisorbed the observed free-electron like state was simply identified as a metal surface state [3], whereas in systems where the molecule is chemisorbed the origin of the state is not properly understood [4,5].
In the scanning tunnelling spectroscopy (STS) study by Kanazawa et al [5] an anisotropic free-electron-like state (AFES) was observed in a p(2×4) monolayer of glycinate anions on the copper (100) surface just above the Fermi energy. From an analysis of autocorrelations of the differential conductance (dI / dV) images, they were able to show that the level of dispersion in the [110] and [110] directions differs by an order of magnitude. Since they did not observe a surface state on the clean surface in the dI / dV spectra, they suggested that the state originated from an interaction between the glycinate monolayer and the copper surface and its anisotropic dispersion from the different strength of interaction between neighbouring radicals in the two directions. There is a definite need to carry out electronic structure calculations to clarify the origin of this state.
In this communication we present the results of density functional theory (DFT) calculations on the p(2×4) glycinate monolayer and show that the origin of the AFES is a copper Shockley surface state (SS). Although Kanazawa et al did not observe a SS near the Fermi energy on the bare Cu(100) surface [5] a SS has been identified near the Fermi energy at the surface Brillouin zone (SBZ) boundary [6,7,8,9]. We show that the observation of a AFES is due to enhanced tunnelling into the SS, mediated by the lowest unoccupied molecular orbitals of the glycinate ions and by the backfolding of the SS to the Γ point in the SBZ for the p(2×4) structure.
Glycine (NH2CH2COOH) is the simplest amino acid and its adsorption geometry on the Cu(100) surface is well understood following several studies in the last ten years with scanning tunnelling microscopy (STM) [5,10,11], photoelectron diffraction [12] and DFT calculations [13]. The glycine molecule loses a hydrogen atom on adsorption becoming a glycinate anion (NH2CH2COO-) which then binds to the copper surface through the nitrogen atom and two oxygen atoms in a tridentate fashion. The p(2×4) structure [5,11] has alternating rows of the left and right enantiomers of glycinate ions, the rows propagating along the close-packed rows of copper atoms (see figure 1). In addition to the heterochiral p(2×4) structure a homochiral c(2×4) [10,11,14] structure is also formed at the same temperature on the Cu(100) surface.
Density functional theory (DFT) calculations were carried out using the plane-wave based VASP code [15]. Calculations were made using the projector augmented wave method [16] and the generalized gradient approximation for the exchange-correlation functional [17]. Glycinate ions were placed on a six layer Cu(100) slab in a 5.14×10.28×25 Å3 supercell. A full geometry relaxation was carried out with a plane wave cutoff of 400 eV on a 8×4×1 k-point grid until the forces acting on the ions were smaller in magnitude than 0.01 eV/Å. The atoms in the top three copper layers were also allowed to relax. Calculations were also performed with a 12 layer bare and adsorbate-covered copper slab to ensure that effects caused by the finite thickness of the slab were not important.
Following the commonly used Tersoff-Hamann approximation [18] the differential conductance measured in STS experiments was approximated using the local density of states (LDOS) calculated at the position of the tip apex, which was set at 7 Å from the top layer of copper atoms. Once the relaxed geometry had been found, the local density of states (LDOS) and molecular orbital projected density of states (MO-PDOS) were calculated on the original k-point grid and in lines from the Γ point to both the X and Y points in the SBZ (figure 1(c)). These calculations used a charge density calculated self-consistently on a 16×8×1 k-point grid. The MO-PDOS was calculated by projecting the density of states of the full system onto the wavefunctions for the isolated monolayer in the same supercell at each k-point.
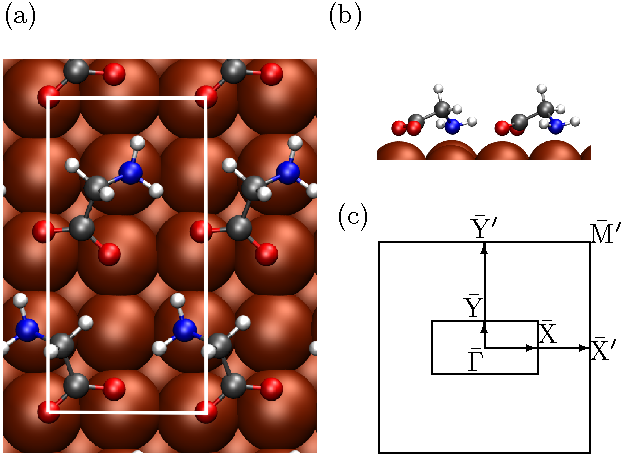
Figure 1 shows the adsorption geometry of the glycinate molecules following geometry relaxation from one close to that found by Mae and Morikawa [13] for the p(2×4) structure. The resulting Cu-N bond length of 2.10 Å and the Cu-O bond lengths of 2.08 Å and 2.18 Å are in good agreement with previous theoretical [13] and experimental [12] findings.
Figure 1: Calculated adsorption geometry of glycinate molecules on Cu(100) (a) top view and (b) side view. The 2×4 unit cell is shown from above in white. (c) Labels for high symmetry points in the square SBZ of the Cu(100) surface and the rectangular SBZ of the p(2×4) glycinate system. The Γ-X direction corresponds to the direction in real space labelled [110] by Kanazawa et al [5], and Γ-Y the [110] direction. The X′ and Y′ points in the substrate SBZ fold back to the Γ point in the monolayer SBZ.
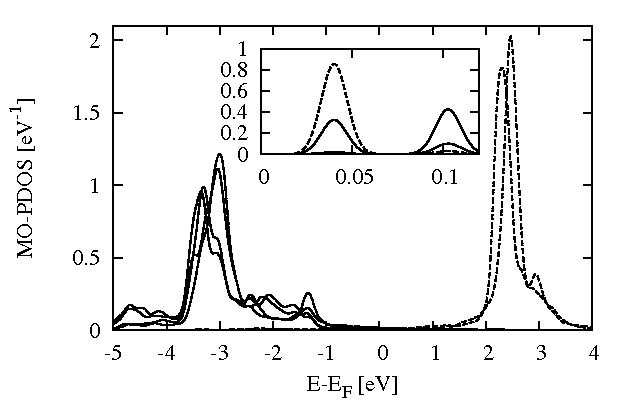
The electronic structure of the molecular monolayer was investigated by calculating the MO-PDOS of the adsorbed system along with the band structure of an isolated layer of neutral glycinate radicals in the same geometry as on the copper surface. The isolated layer is found to have a group of four closely lying bands which are only partially occupied. They are formed by the overlap of molecular orbitals on neighbouring molecules and have a width of approximately 0.4 eV across the SBZ. Projection of the DOS of the adsorbed system onto these partially occupied bands in the isolated glycinate layer shows that the bands become fully occupied in the presence of the copper surface forming negatively charged glycinate anions (figure 2). This is in agreement with previous studies [5,11,12,19]. The glycinate states are considerably broadened upon adsorption reflecting a strong bonding interaction between the glycinate anions and the copper surface. Although bands are formed due to interactions between neighbouring molecules they lie too low in energy and do not have the correct dispersion behaviour to explain the observed AFES.
Figure 2: Plot of the MO-PDOS for the adsorbed system integrated over the whole SBZ. Solid lines are projected onto the bands which are partially occupied in the isolated layer of glycinate radicals, dashed lines onto the first two unoccupied bands. As there are two molecules per unit cell, bands appear in pairs. States are broadened using Gaussians of width 0.1 eV. Inset shows the MO-PDOS for the same system at the Γ point near the Fermi energy broadened with Gaussians which are narrower (width 0.01 eV) and taller for better resolution.
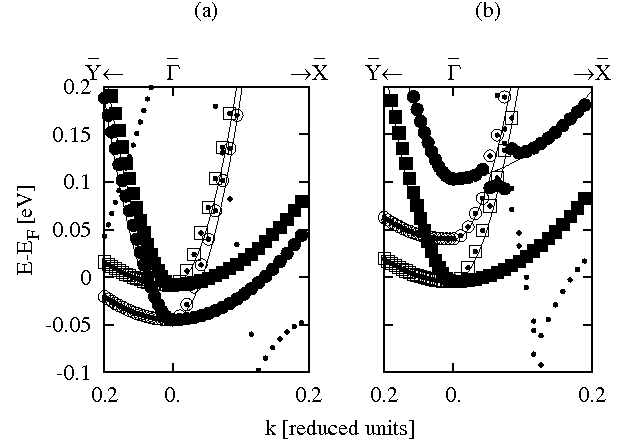
Calculations show that the SS of the bare copper slab has a similar energy and anisotropic dispersion to the observed AFES. The SSs are clearly observable in the calculated band structure of the bare copper slab shown in figure 3(a). The SSs on the top of the slab are 37 meV lower than those on the bottom because the top layers are relaxed. Two degenerate SSs are found on each surface of the bare slab; one arising from the X′ point and one from the equivalent Y′ point in the (1×1) unit cell. These states are both folded back to the Γ point in the p(2×4) unit cell and can be distinguished by comparing their dispersion in the Γ-X (|| [110] || Γ′-X′) and Γ-Y (|| [110] || X′-M′) directions. The SS arising from the X′ point has a smaller effective mass in the Γ-X direction and that arising from the Y′ point has a smaller effective mass in the Γ-Y direction. The calculated dispersion of the SS originating from the X′ point reproduces closely the observed parabolic dispersion with an effective mass of 0.067±0.01 me in the [110] direction measured by Kevan [6] in an angle resolved photoemission study. The calculated SS energy of 45 meV below the Fermi energy at the Γ point is also in good agreement with the measured onset of 58±0.05 meV below the Fermi energy [6]. The effective mass of 0.067±0.01 me in the [110] direction of the SS is close the measured effective mass of 0.061 me of the AFES in the same direction. Furthermore, the same calculated SS in figure 3(a) also shows a parabolic-like dispersion in the [110] direction in close agreement with the measured dispersion of the AFES in the same surface direction with an effective mass of 0.61 me.
A comparison of the calculated band structures in figure 3(a) and (b) shows that the SSs of the bare copper slab preserve their character in the presence of the glycine anions and experience only small energy shifts. Upon adsorption, the degeneracy of the SSs at the X′ and the Y′ points is lifted and their energies are shifted up by 85 meV and 148 meV, respectively. The energies of the SSs from both the X′ and Y′ points on the bottom side of the slab remain constant within a few meV and are degenerate, showing that the slab is thick enough to prevent interactions between the monolayer and the SSs at the bottom of the slab. The origin of these adsorption-induced energy shifts of the SSs was investigated by calculating the SS energies of the frozen bare slab where copper atoms were held in the geometry they had in the presence the glycinate anions. In this case the SSs at the X′ and Y′ points are only shifted by 48 meV and 30 meV respectively. Although part of the energy shifts are due to the adsorbate-induced changes in geometry of the copper surface, large parts of the energy shifts are only observable in the presence of the glycinate anions. A substantial part of the shift in energy must therefore be due to electrostatic effects and direct electronic interaction between the SSs and the glycinate anions states. The inset in figure 2 show that there is a small interaction with molecular states but figure 3 shows that this is not sufficiently large to change the dispersion of the SSs close to the Γ point.
Figure 3: Bandstructure in the p(2×4) unit cell of (a) the bare and (b) the glycine covered Cu(100) slab. k-points are plotted in reduced units to one fifth of the distance to the zone boundary. Squares(circles) represent states from the bottom(top) of the slab. Open(closed) symbols have copper px(py) character. The remaining dots represent other states. The solid curves show the parabolic dispersion for the observed AFES [5].
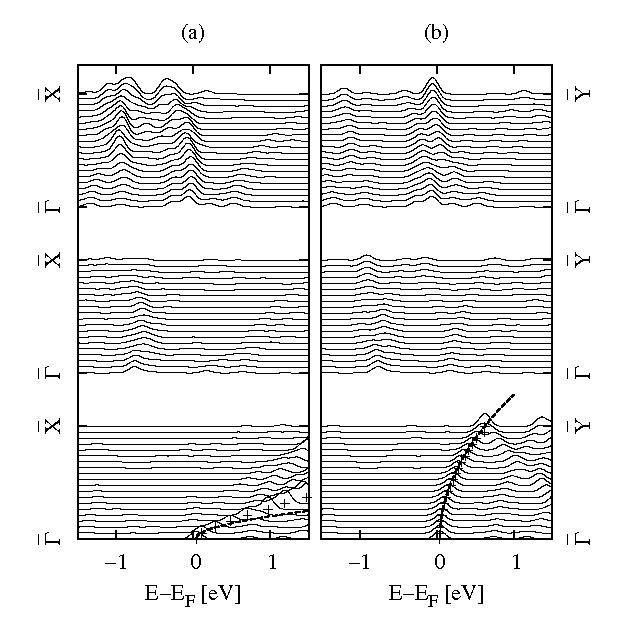
The wave-vector resolved LDOS of the glycinate covered surface in figure 4 shows that only the AFES originating from SS at the X′ gives rise to a peak in the LDOS and corresponds to the observed AFES. The dispersion of this peak agrees very well with the experimentally observed parabolic dispersion with effective masses of 0.061 me and 0.61 me in the [110] and the [110] directions, respectively, although the experimentally observed onset of the peak was at 130 meV above the Fermi energy, slightly higher than the calculated value of 40 meV. Furthermore, that the calculated AFES peak in the LDOS arises solely from the molecular states in the monolayer is ruled out by the behaviour of the LDOS when changing the distance of the glycinate ion monolayer from the copper surface by 0.5 Å and 1 Å without subsequent relaxation of the geometry. The AFES peak in the LDOS is seen to get gradually smaller as the glycinate ions are withdrawn from the surface (figure 4). A peak due to tunnelling through a purely molecular state would get taller as the molecules were brought closer to the tip. This behaviour can be seen by the formation of peaks at the Fermi energy which arise from the previously discussed partially occupied bands of the isolated overlayer. The AFES peak gets smaller as the molecules are withdrawn which is neither consistent with tunnelling through a purely molecular state nor directly into an unperturbed copper surface state. If the peak were only due to direct tunnelling into the copper substrate then the height of the peak would be constant.
Figure 4: Plots of the wavevector resolved LDOS above the copper surface along a line of k-points from Γ to (a) X and (b) Y. Data from neighbouring k-points have been displaced vertically. The lower set of curves are for the fully relaxed adsorbate geometry, the middle and upper set with the glycinate ions displaced 0.5 Å and 1.0 Å away from the copper surface. For the lower set of curves, crosses mark the position of the top of the AFES peak at its base and the dashed curves are parabolic dispersions with the effective masses observed by Kanazawa et al [5].
Since the AFES originating from the SS could only be observed on the glycinate covered surface but not on the bare surface using STS [5], the tunnelling into the SS must be enhanced by the monolayer. Kanazawa et al observed a peak in the dI / dV spectrum over the glycinate monolayer with the steep onset and gradual decay with increasing bias characteristic of a dispersive state, but no such peak was observed in the dI / dV spectrum over the clean copper surface. Calculations of the LDOS at 7 Å above the bare copper slab show no contribution due to the SSs, only contributions from bulk states around the Γ point. On the bare surface the SSs at the X′ and Y′ points decay much more rapidly into the vacuum region than states with the same energy around Γ point and the tunnelling is dominated by these latter states.
Back folding of the zone boundary SSs to the zone centre is not sufficient to make them observable in tunnelling spectrocopy. Although both the X′ and Y′ points are folded back to the Γ point, only the SS arising from the X′ point is responsible for the AFES in the LDOS. This is in spite of the fact that a larger energy shift is found for the SS arising from the Y′ point, indicating a stronger interaction with the glycinate anions. This result agrees nicely with the observation in the STS experiments [5] that the AFES has a smaller effective mass in the Γ-X direction than in the Γ-Y direction. The SS arising from the Y′ point would have a higher effective mass in Γ-X direction than in the Γ-Y direction. That only the SS from the X′ point contributes to the LDOS is consistent with the suggestion that the LUMO of the glycinate anions plays an important rôle in the tunnelling into the SS. The separate contributions to the MO-PDOS from different k-points in the Γ-X and Γ-Y directions show that the SS giving rise to the AFES in the LDOS has non-zero overlap with several of the glycinate anion states. The largest overlap is with the first fully unoccupied band in the isolated monolayer, the LUMOs of the glycinate anions (inset of figure 2). In contrast, the SS arising from the Y′ point has no discernible overlap with these molecular orbitals. This is not simply due to the symmetry of the p(2×4) system. The monolayer has only one non-trivial symmetry operation: a combined translation by two copper lattice spacings in the Γ-Y direction and a reflection. Because of the predominant p character of the SSs at the X′ and Y′ points, they are both even under this symmetry operation. We suggest that there is a better match of the local symmetry between the LUMO and the SS arising from the X′ point than that arising from the Y′ point. The overlap of only the SS arising from the X′ point with the LUMO suggests that this small overlap is sufficient for tunnelling mediated by the LUMO into this substrate SS beneath the molecules.
The AFES was not observed on the alternative c(2×4) structure [14], and as calculations show that peaks in the LDOS originating from the SS, though present, are an order of magnitude smaller than for the p(2×4) structure, we have concentrated on the p(2×4) structure.
In conclusion, we have shown from our density functional calculations that an organic molecular monolayer can dramatically enhance the tunnelling into a metal surface state that is otherwise not observed in scanning tunnelling spectroscopy (STS). This finding is of importance in understanding the origin of free-electron like states in tunnelling through molecular monolayers on surfaces. These states are not necessarily due to the formation of bands within the molecular monolayer itself and may be mainly localized in the substrate.
The authors would like to thank the Marie Curie Research Training Network PRAIRIES, contract MRTN-CT-2006-035810, and the Swedish Research Council (VR) for financial support and MSD for funding of postdoctoral fellowship and computational resources by the the University of Liverpool.