


|
Decoding the proton veil around lysosomes Massimiliano Stagi
AbstractNow available in Nature Cell Biology: my News & Views article titled “Decoding the proton veil around lysosomes.” This piece was inspired by an excellent study, which can be found here: https://lnkd.in/exeesr76. I hope it provokes thought and discussion among readers, just as it did for me. You can read the News & Views article here: https://lnkd.in/egzYajpc. |

|
RpH-ILV: Probe for lysosomal pH and acute LLOMe-induced membrane permeabilization in cell lines and Drosophila Izaak J. Cheetham-Wilkinson, Bhavya Sivalingam, Chloe Flitton, Franziska Flottmann, Luisa Vehling, Maik Drechsler, Marija Stojchevska, Andrea Raimondi, Achim Paululat, Florian Fröhlich, Laura E. Swan(#), Massimiliano Stagi(#)
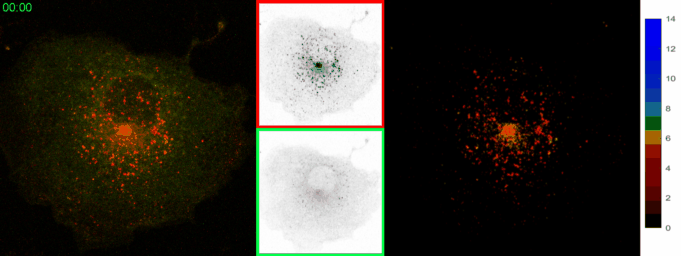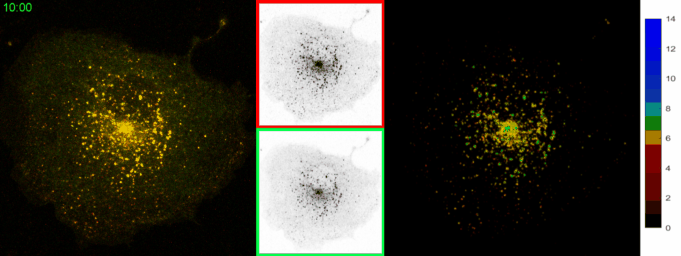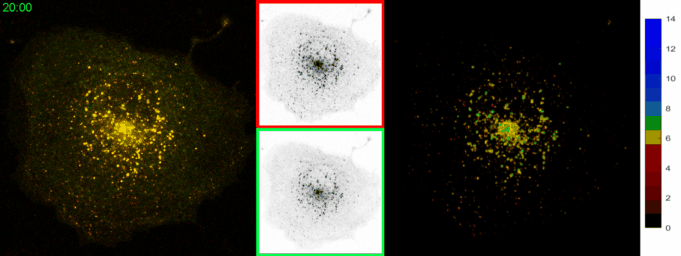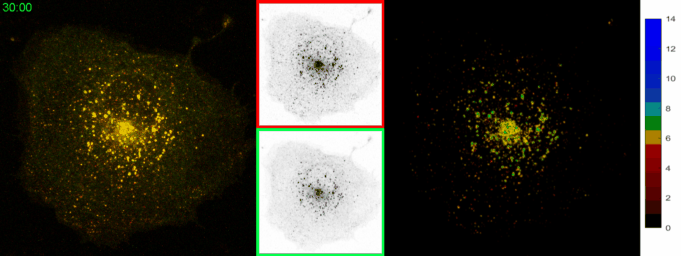
AbstractLysosomal pH dysregulation is a critical element of the pathophysiology of neurodegenerative diseases, cancers, and lysosomal storage disorders (LSDs). To study the role of lysosomes in pathophysiology, probes to analyze lysosomal size, positioning, and pH are indispensable tools. Here, we developed and characterized a ratiometric genetically encoded lysosomal pH probe, RpH-ILV, targeted to a subpopulation of lysosomal intraluminal vesicles. This subpopulation behaves similarly to the general population of LAMP1-positive vesicles in terms of pH response to pharmacological stresses. In addition, RpH-ILV, which is trafficked to the lysosome via a different cytosolic motif than our previous ratiometric sensor, RpH-LAMP1, is well tolerated by the model organism Drosophila melanogaster, exhibits minimal plasma membrane fluorescence, and reveals sensitivity to the lysosomal damaging agent LLOMe, adding a valuable tool to our repertoire of lysosomal pH sensors. |
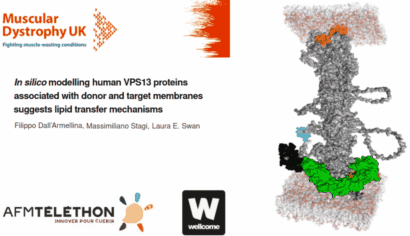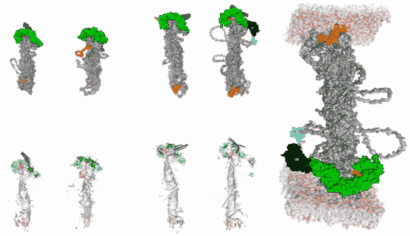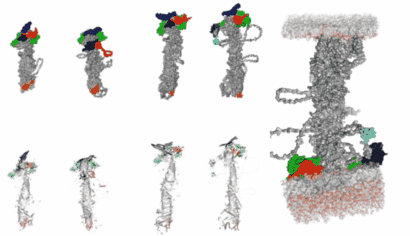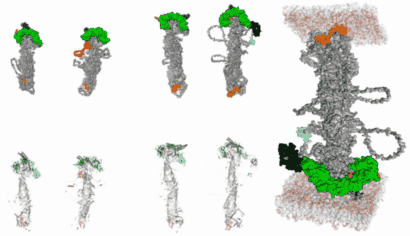 |
In silico modelling human VPS13 proteins associated with donor and target membranes suggests lipid transfer mechanisms. Filippo Dall'Armellina Stagi, M.,Laura E. Swan
AbstractThe VPS13 protein family constitutes a novel class of bridge-like lipid transferases. Autosomal recessive inheritance of mutations in VPS13 genes is associated with the development of neurodegenerative diseases in humans. Bioinformatic approaches previously recognised the domain architecture of these proteins. In this study, we model the first ever full-length structures of the four human homologs VPS13A, VPS13B, VPS13C, and VPS13D in association with model membranes, to investigate their lipid transfer ability and potential structural association with membrane leaflets. We analyse the evolutionary conservation and physicochemical properties of these proteins, focusing on conserved C-terminal amphipathic helices that disturb organelle surfaces and that, adjoined, resemble a traditional Venetian gondola. The gondola domains share significant structural homology with lipid droplet surface-binding proteins. We introduce in silico protein-membrane models displaying the mode of association of VPS13A, VPS13B, VPS13C, and VPS13D to donor and target membranes, and present potential models of action for protein-mediated lipid transfer. |
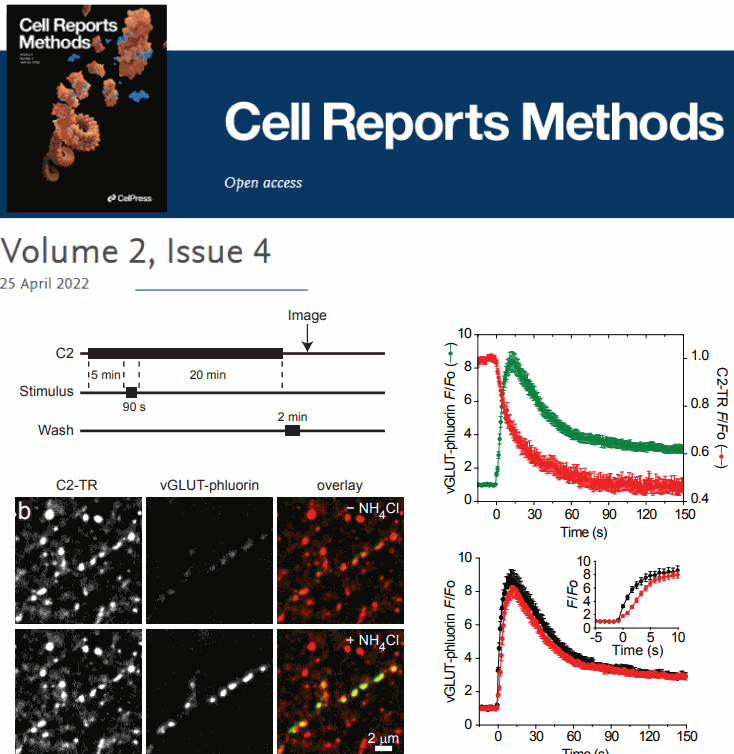 |
Multimodal Imaging of Synaptic Vesicles with a Single Probe. Seong J.An (*), Massimiliano Stagi(*) ,Travis J.Gould, YumeiWu, MichaelMlodzianoski, FelixRivera-Molina, DerekToomre, Stephen M.Strittmatter, PietroDe Camilli, JoergBewersdorf, DavidZenisek
AbstractA complete understanding of synaptic vesicle recycling requires the use of multiple microscopy methods to obtain complementary information. However, many currently available probes are limited to a specific microscopy modality, which necessitates the use of multiple probes and labeling paradigms. Given the complexity of vesicle populations and recycling pathways, having new single vesicle probes that could be used for multiple microscopy techniques would complement existing sets of tools for studying vesicle function. Here we present a novel probe based on the membrane-binding C2 domain of cytosolic phospholipase A2 (cPLA2) that fulfills this need. By conjugating the C2 domain with different detectable tags, we demonstrate that a single, modular probe can allow synaptic vesicles to be imaged at multiple levels of spatial and temporal resolution. |
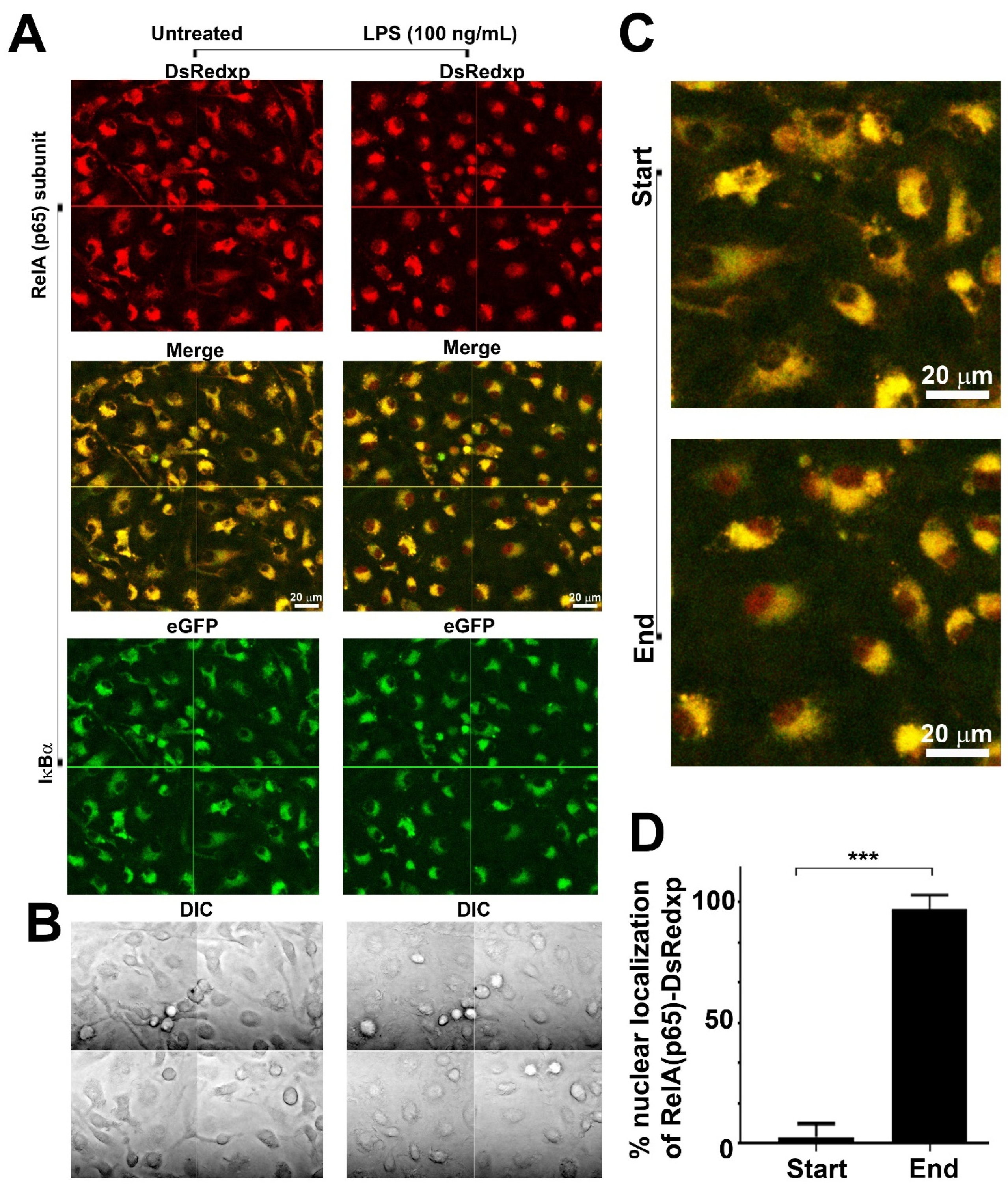 |
Effects of Human RelA Transgene on Murine Macrophage Inflammatory Responses Papoutsopoulou, S., Morris, L., Bayliff, A., Mair, T., England, H., Stagi, M., . . . Campbell, B. J.
AbstractThe NFκB transcription factors are major regulators of innate immune responses, and NFκB signal pathway dysregulation is linked to inflammatory disease. Here, we utilised bone marrow-derived macrophages from the p65-DsRedxp/IκBα-eGFP transgenic strain to study the functional implication of xenogeneic (human) RelA(p65) protein introduced into the mouse genome. Confocal imaging showed that human RelA is expressed in the cells and can translocate to the nucleus following activation of Toll-like receptor 4. RNA sequencing of lipid A-stimulated macrophages, revealed that human RelA impacts on murine gene transcription, affecting both non-NFκB and NFκB target genes, including immediate-early and late response genes, e.g., Fos and Cxcl10. Validation experiments on NFκB targets revealed markedly reduced mRNA levels, but similar kinetic profiles in transgenic cells compared to wild-type. Enrichment pathway analysis of differentially expressed genes revealed interferon and cytokine signaling were affected. These immune response pathways were also affected in macrophages treated with tumor necrosis factor. Data suggests that the presence of xenogeneic RelA protein likely has inhibitory activity, altering specific transcriptional profiles of key molecules involved in immune responses. It is therefore essential that this information be taken into consideration when designing and interpreting future experiments using this transgenic strain |
.jpg)  |
Live imaging of intra-lysosome pH in cell lines and primary neuronal culture using a novel genetically encoded biosensor. Amy H. Ponsford , Thomas A. Ryan , Andrea Raimondi , Emanuele Cocucci, Susanne A. Wycislo , Florian Froehlich(#), Laura E. Swan (#), Massimiliano Stagi (#)
AbstractDisorders of lysosomal physiology have increasingly been found to underlie the pathology of a rapidly growing cast of neurodevelopmental disorders and sporadic diseases of aging. One cardinal aspect of lysosomal (dys)function is lysosomal acidification in which defects trigger lysosomal stress signaling and defects in proteolytic capacity. We have developed a genetically encoded ratiometric probe to measure lysosomal pH coupled with a purification tag to efficiently purify lysosomes for both proteomic and in vitro evaluation of their function. Using our probe, we showed that lysosomal pH is remarkably stable over a period of days in a variety of cell types. Additionally, this probe can be used to determine that lysosomal stress signaling via TFEB is uncoupled from gross changes in lysosomal pH. Finally, we demonstrated that while overexpression of ARL8B GTPase causes striking alkalinization of peripheral lysosomes in HEK293T cells, peripheral lysosomes per se are no less acidic than juxtanuclear lysosomes in our cell lines. |
 |
Lowe Syndrome-linked endocytic adaptors direct membrane cycling kinetics with OCRL in Dictyostelium discoideum. Alexandre Luscher, Florian Fruhlich, Caroline Barisch, Clare Littlewood, Joe Metcalfe, Florence Leuba, Anita Palma, Michelle Pirruccello, Gianni Cesareni, Massimiliano Stagi , Tobias C. Walther, Thierry Soldati, Pietro De Camilli, and Laura E. Swan
AbstractMutations of the inositol 5-phosphatase OCRL cause Lowe Syndrome (LS), characterized by congenital cataract, low IQ and defective kidney proximal tubule resorption. A key subset of LS mutants abolishes OCRL's interactions with endocytic adaptors containing F&H peptide motifs. Converging unbiased methods examining human peptides and the unicellular phagocytic organism Dictyostelium discoideum, reveal that, like OCRL, the Dictyostelium OCRL orthologue Dd5P4 binds two proteins closely related to the F&H proteins APPL1 and Ses1/2 (also referred to as IPIP27A/B). In addition, a novel conserved F&H interactor was identified, GxcU (in Dictyostelium) and the Cdc42-GEF Frabin (in human cells). Examining these proteins in Dictyostelium discoideum, we find that, like OCRL, Dd5P4 acts at well-conserved and physically distinct endocytic stations. Dd5P4 functions in coordination with F&H proteins to control membrane deformation at multiple stages of endocytosis, and suppresses GxcU-mediated activity during fluid-phase micropinocytosis. We also reveal that OCRL/Dd5P4 acts at the contractile vacuole, an exocytic osmoregulatory organelle. We propose F&H peptide-containing proteins may be key modifiers of LS phenotypes. |
|
|
Mitochondrial respiratory chain deficiency inhibits lysosomal hydrolysis. Lorena Fernandez-Mosquera, King F. Yambire, Renata Couto, Leonardo Pereyra, Kamil Pabis, Amy H. Ponsford, C�tia V. Diogo1, Massimiliano Stagi, Ira Milosevic, Nuno Raimundo
AbstractMitochondria are key organelles for cellular metabolism, and regulate several processes including cell death and autophagy. Here, we show that mitochondrial respiratory chain (RC) deficiency deactivates AMPactivated protein kinase (AMPK, a key regulator of energy homeostasis) signaling in tissue and in cultured cells. The deactivation of AMPK in RC-deficiency is due to increased expression of the AMPK-inhibiting protein folliculin. AMPK is found to be necessary for basal lysosomal function, and AMPK deactivation in RC-deficiency inhibits lysosomal function by decreasing the activity of the lysosomal Ca2+ channel Mucolipin-1 (MCOLN1). MCOLN1 is regulated by phosphoinositide kinase PIKfyve and its product PI(3,5)P2, which is also decreased in RC-deficiency. Notably, reactivation of AMPK, in a PIKfyve-dependent manner, or of MCOLN1 in RC deficient cells, restores lysosomal hydrolytic capacity. Building on these data and the literature, we propose that down-regulation of the AMPK-PIKfyve-PI(3,5)P2-MCOLN1 pathway causes lysosomal Ca2+ accumulation and impaired lysosomal catabolism. Besides unveiling a novel role of AMPK in lysosomal function, this study points to the mechanism that links mitochondrial malfunction to impaired lysosomal catabolism, underscoring the importance of AMPK and the complexity of organelle cross-talk in the regulation of cellular homeostasis. |
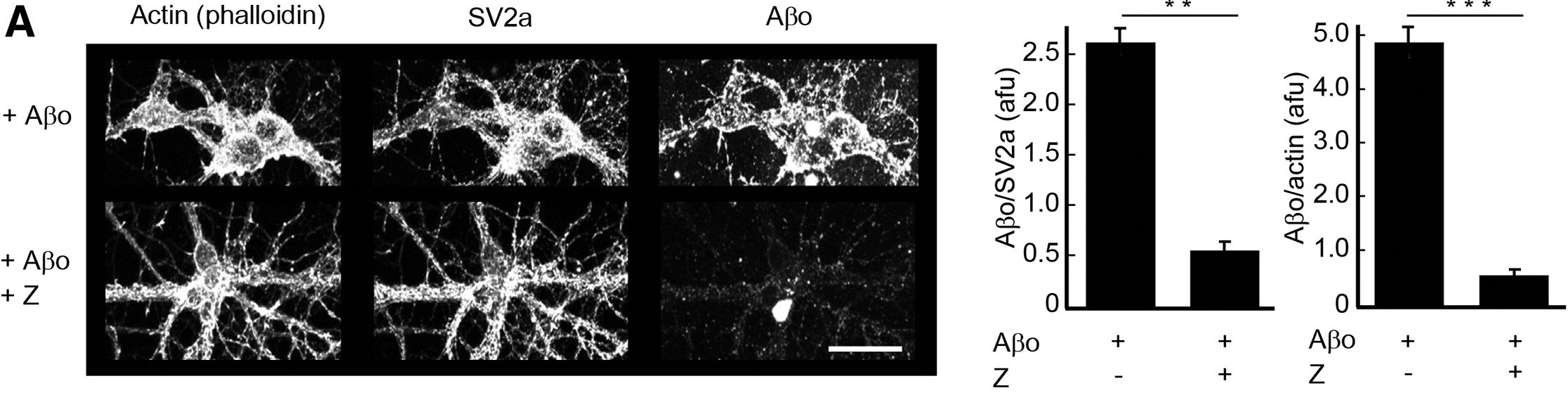 |
Rescue of Transgenic Alzheimer's Pathophysiology by Polymeric Cellular Prion Protein Antagonists. Gunther EC, Smith LM, Kostylev MA, Cox TO, Kaufman AC, Lee S, Folta-Stogniew E, Maynard GD, Um JW,Massimiliano Stagi , Heiss JK, Stoner A, Noble GP, Takahashi H1, Haas LT, Schneekloth JS, Merkel J, Teran C, Naderi ZK, Supattapone S, Strittmatter SM
AbstractCellular prion protein (PrPC) binds the scrapie conformation of PrP (PrPSc) and oligomeric Beta-amyloid peptide (ABetaO) to mediate transmissible spongiform encephalopathy (TSE) and Alzheimer's disease (AD), respectively. We conducted cellular and biochemical screens for compounds blocking PrPC interaction with ABetaO. A polymeric degradant of an antibiotic targets ABetaO binding sites on PrPC with low nanomolar affinity and prevents ABetaO-induced pathophysiology. We then identified a range of negatively charged polymers with specific PrPC affinity in the low to sub-nanomolar range, from both biological (melanin) and synthetic (poly [4-styrenesulfonic acid-co-maleic acid], PSCMA) origin. Association of PSCMA with PrPC prevents ABetaO/PrPC-hydrogel formation, blocks ABetaO binding to neurons, and abrogates PrPSc production by ScN2a cells. We show that oral PSCMA yields effective brain concentrations and rescues APPswe/PS1?E9 transgenic mice from AD-related synapse loss and memory deficits. Thus, an orally active PrPC-directed polymeric agent provides a potential therapeutic approach to address neurodegeneration in AD and TSE. |
 |
Loss of TMEM106B Ameliorates Lysosomal and Frontotemporal Dementia-Related Phenotypes in Progranulin-Deficient Mice Zoe A. Klein, Hideyuki Takahashi, Mengxiao Ma,Massimiliano Stagi , Melissa Zhou, TuKiet T. Lam, Stephen M. Strittmatter
AbstractProgranulin (GRN) and TMEM106B are associated with several common neurodegenerative disorders including frontotemporal lobar degeneration (FTLD). A TMEM106B variant modifies GRN-associated FTLD risk. However, their functional relationship in vivo and the mechanisms underlying the risk modification remain unclear. Here, using transcriptomic and proteomic analyses with Grn-/- and Tmem106b-/- mice, we show that, while multiple lysosomal enzymes are increased in Grn-/- brain at both transcriptional and protein levels, TMEM106B deficiency causes reduction in several lysosomal enzymes. Remarkably, Tmem106b deletion from Grn-/- mice normalizes lysosomal protein levels and rescues FTLD-related behavioral abnormalities and retinal degeneration without improving lipofuscin, C1q, and microglial accumulation. Mechanistically, TMEM106B binds vacuolar-ATPase accessory protein 1 (AP1). TMEM106B deficiency reduces vacuolar-ATPase AP1 and V0 subunits, impairing lysosomal acidification and normalizing lysosomal protein levels in Grn-/- neurons. Thus, Grn and Tmem106b genes have opposite effects on lysosomal enzyme levels, and their interaction determines the extent of neurodegeneration. |
|
Co-culture synaptogenic assay: A new look at fluorescence reporters and technological devices (Chapter) de Arce, K. P.(*), & Stagi M.(#)
AbstractThe mechanism underlying the differentiation of pre- and postsynaptic specifications involves the sequential and dynamic recruitment of specific molecules coordinated by bidirectional signaling across the synaptic cleft. In this chapter, we describe the co-culture assay, a useful method to evaluate cell-surface molecules through its ability to promote the recruitment of proteins required for synapse structure and function. The versatility of this simple and reliable method is illustrated by the wide variety of applications ranging from analysis of synaptogenic activity to evaluation of soluble compounds with therapeutic potential. In addition, we provide a framework to enable the co-culture assay as a tool for high-throughput studies, thereby improving the efficiency and sensitivity of this classic method in neuroscience. |
 |
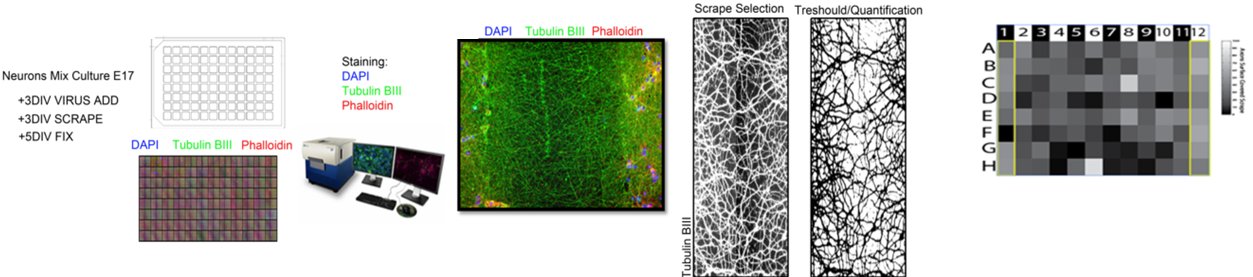 |
Gene-Silencing Screen for Mammalian Axon Regeneration Identifies Inpp5f (Sac2) as an Endogenous Suppressor of Repair after Spinal Cord Injury Y. Zou (*), M .Stagi (*), X. Wang, K. Yigitkanli, C. S. Siegel, F. Nakatsu, W. B. J. Cafferty, and S. M. Strittmatter
AbstractAxonal growth and neuronal rewiring facilitate functional recovery after spinal cord injury. Known interventions that promote neural repair remain limited in their functional efficacy. To understand genetic determinants of mammalian CNS axon regeneration, we completed an unbiased RNAi gene-silencing screen across most phosphatases in the genome. We identified one known and 17 previously unknown phosphatase suppressors of injury-induced CNS axon growth. Silencing Inpp5f (Sac2) leads to robust enhancement of axon regeneration and growth cone reformation. Results from cultured Inpp5f−/− neurons confirm lentiviral shRNA results from the screen. Consistent with the nonoverlapping substrate specificity between Inpp5f and PTEN, rapamycin does not block enhanced regeneration in Inpp5f−/− neurons, implicating mechanisms independent of the PI3K/AKT/mTOR pathway. Inpp5f−/− mice develop normally, but show enhanced anatomical and functional recovery after mid-thoracic dorsal hemisection injury. More serotonergic axons sprout and/or regenerate caudal to the lesion level, and greater numbers of corticospinal tract axons sprout rostral to the lesion. Functionally, Inpp5f-null mice exhibit enhanced recovery of motor functions in both open-field and rotarod tests. This study demonstrates the potential of an unbiased high-throughput functional screen to identify endogenous suppressors of CNS axon growth after injury, and reveals Inpp5f (Sac2) as a novel suppressor of CNS axon repair after spinal cord injury. |
|
Low cost production of 3D-printed devices and electrostimulation chambers for the culture of primary neurons. Wardyna J.D., Sandersona C, Swana L.E. , Stagi M.(*)
AbstractThe analysis of primary neurons is a basic requirement for many areas of neurobiology. However, the range of commercial systems available for culturing primary neurons is functionally limiting, and the expense of these devices is a barrier to both exploratory and large-scale studies. This is especially relevant as primary neurons often require unusual geometries and specialised coatings for optimum growth. Fortunately, the recent revolution in 3D printing offers the possibility to generate customised devices, which can support neuronal growth and constrain neurons in defined paths, thereby enabling many aspects of neuronal physiology to be studied with relative ease. In this article, we provide a detailed description of the system hardware and software required to produce affordable 3D-printed culture devices, which are also compatible with live-cell imaging. In addition, we also describe how to use these devices to grow and stimulate neurons within geometrically constrained compartments and provide examples to illustrate the practical utility and potential that these protocols offer for many aspects of experimental neurobiology. |
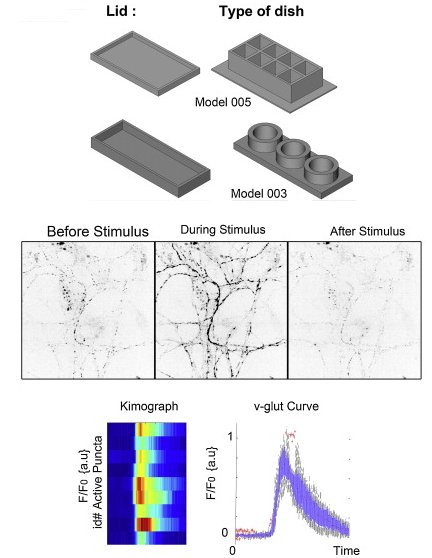 |
 |
Lysosome size, motility and stress response regulated by fronto-temporal dementia modifier TMEM106B. Stagi M, Zoe A. Klein, Travis J. Gould, Joerg Bewersdorf, Strittmatter SM.
AbstractFronto-temporal lobar degeneration with TDP-43 (FTLD-TDP) is a fatal neurodegeneration. TMEM106B variants are linked to FTLD-TDP risk, and TMEM106B is lysosomal. Here, we focus on neuronal TMEM106B, and demonstrate co-localization and traffic with lysosomal LAMP-1. pH-sensitive reporters demonstrate that the TMEM106B C-terminus is lumenal. The TMEM106B N-terminus interacts with endosomal adaptors and other TMEM106 proteins. TMEM106B knockdown reduces neuronal lysosomal number and diameter by STED microscopy, and overexpression enlarges LAMP-positive structures. Reduction of TMEM106B increases axonally transported lysosomes, while TMEM106B elevation inhibits transport and yields large lysosomes in the soma. TMEM106B overexpression alters lysosomal stress signaling, causing a translocation of the mTOR-sensitive transcription factor, TFEB, to neuronal nuclei. TMEM106B loss-of-function delays TFEB translocation after Torin-1-induced stress. Enlarged TMEM106B-overexpressing lysosomes maintain organelle integrity longer after lysosomal photodamage than do control lysosomes, while small TMEM106B-knockdown lysosomes are more sensitive to illumination. Thus, neuronal TMEM106B plays a central role in regulating lysosomal size, motility and responsiveness to stress, highlighting the possible role of lysosomal biology in FTLD-TDP. |
|
Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer ABetaO oligomer bound to cellular prion protein. Um JW, Kaufman AC, Kostylev M, Heiss JK, Stagi M, Takahashi H, Kerrisk ME, Vortmeyer A, Wisniewski T, Koleske AJ, Gunther EC, Nygaard HB, Strittmatter SM.
AbstractSoluble amyloid-Beta oligomers (ABetao) trigger Alzheimer's disease (AD) pathophysiology and bind with high affinity to cellular prion protein (PrP(C)). At the postsynaptic density (PSD), extracellular ABetao bound to lipid-anchored PrP(C) activates intracellular Fyn kinase to disrupt synapses. Here, we screened transmembrane PSD proteins heterologously for the ability to couple ABetao-PrP(C) with Fyn. Only coexpression of the metabotropic glutamate receptor, mGluR5, allowed PrP(C)-bound ABetao to activate Fyn. PrP(C) and mGluR5 interact physically, and cytoplasmic Fyn forms a complex with mGluR5. Abetao-PrP(C) generates mGluR5-mediated increases of intracellular calcium in Xenopus oocytes and in neurons, and the latter is also driven by human AD brain extracts. In addition, signaling by ABetao-PrP(C)-mGluR5 complexes mediates eEF2 phosphorylation and dendritic spine loss. For mice expressing familial AD transgenes, mGluR5 antagonism reverses deficits in learning, memory, and synapse density. Thus, ABetao-PrP(C) complexes at the neuronal surface activate mGluR5 to disrupt neuronal function. |
 |
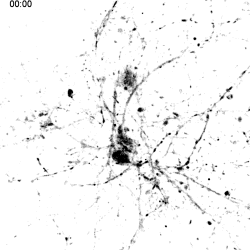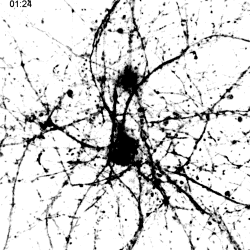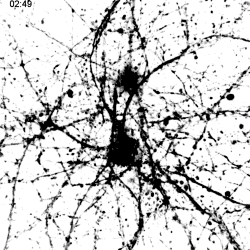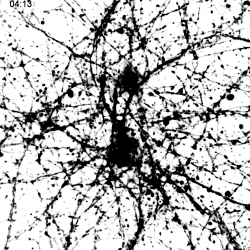 |
Alzheimer amyloid-ABetaO oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Um JW, Nygaard HB, Heiss JK, Kostylev MA, Stagi M, Vortmeyer A, Wisniewski T, Gunther EC, Strittmatter SM.
AbstractAmyloid-beta (ABeta) oligomers are thought to trigger Alzheimer's disease pathophysiology. Cellular prion protein (PrP(C)) selectively binds oligomeric ABeta and can mediate Alzheimer's disease-related phenotypes. We examined the specificity, distribution and signaling of ABeta-PrP(C) complexes, seeking to understand how they might alter the function of NMDA receptors (NMDARs) in neurons. PrP(C) is enriched in postsynaptic densities, and ABeta-PrP(C) interaction leads to Fyn kinase activation. Soluble ABeta assemblies derived from the brains of individuals with Alzheimer's disease interacted with PrP(C) to activate Fyn. ABeta engagement of PrP(C)-Fyn signaling yielded phosphorylation of the NR2B subunit of NMDARs, which was coupled to an initial increase and then a loss of surface NMDARs. ABeta-induced dendritic spine loss and lactate dehydrogenase release required both PrP(C) and Fyn, and human familial Alzheimer's disease transgene-induced convulsive seizures did not occur in mice lacking PrP(C). These results delineate an ABeta oligomer signal transduction pathway that requires PrP(C) and Fyn to alter synaptic function, with deleterious consequences in Alzheimer's disease. |
|
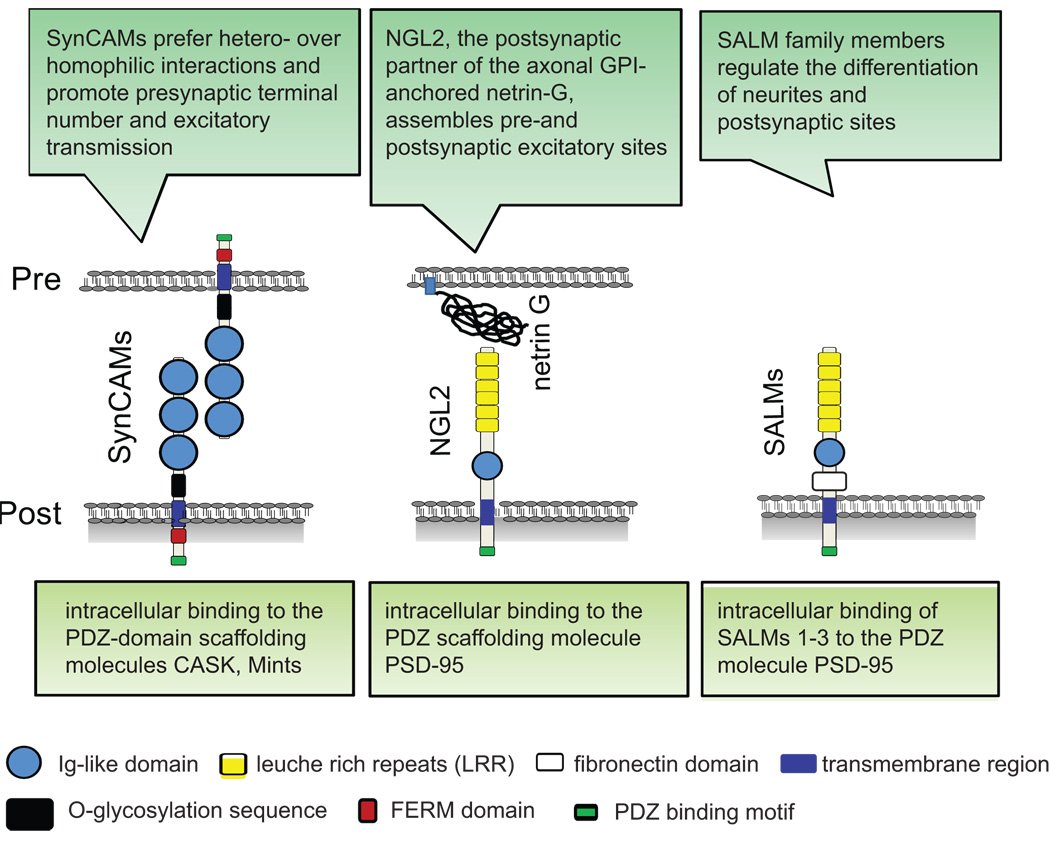
Lateral assembly of the immunoglobulin protein SynCAM 1 controls its adhesive function and instructs synapse formation. * = Equal contribution
Fogel AI*, Stagi M* , TPerez de Arce K*, Biederer T.
AbstractSynapses are specialized adhesion sites between neurons that are connected by protein complexes spanning the synaptic cleft. These trans-synaptic interactions can organize synapse formation, but their macromolecular properties and effects on synaptic morphology remain incompletely understood. Here, we demonstrate that the synaptic cell adhesion molecule SynCAM 1 self-assembles laterally via its extracellular, membrane-proximal immunoglobulin (Ig) domains 2 and 3. This cis oligomerization generates SynCAM oligomers with increased adhesive capacity and instructs the interactions of this molecule across the nascent and mature synaptic cleft. In immature neurons, cis assembly promotes the adhesive clustering of SynCAM 1 at new axo-dendritic contacts. Interfering with the lateral self-assembly of SynCAM 1 in differentiating neurons strongly impairs its synaptogenic activity. At later stages, the lateral oligomerization of SynCAM 1 restricts synaptic size, indicating that this adhesion molecule contributes to the structural organization of synapses. These results support that lateral interactions assemble SynCAM complexes within the synaptic cleft to promote synapse induction and modulate their structure. These findings provide novel insights into synapse development and the adhesive mechanisms of Ig superfamily members. |
 Signals immediately after bleaching of Cherry in the marked area.
The increase in SynCAM 1-pH signal after bleaching the SynCAM 1-Ch donor demonstrates FRET |
 |
SynCAM 1 participates in axo-dendritic contact assembly and shapes neuronal growth cones. Stagi M, Fogel AI, Biederer T.
AbstractNeuronal growth cones are highly motile structures that tip developing neurites and explore their surroundings before axo-dendritic contact and synaptogenesis. However, the membrane proteins organizing these processes remain insufficiently understood. Here we identify that the synaptic cell adhesion molecule 1 (SynCAM 1), an immunoglobulin superfamily member, is already expressed in developing neurons and localizes to their growth cones. Upon interaction of growth cones with target neurites, SynCAM 1 rapidly assembles at these contacts to form stable adhesive clusters. Synaptic markers can also be detected at these sites. Addressing the functions of SynCAM 1 in growth cones preceding contact, we determine that it is required and sufficient to restrict the number of active filopodia. Further, SynCAM 1 negatively regulates the morphological complexity of migrating growth cones. Focal adhesion kinase, a binding partner of SynCAM 1, is implicated in its morphogenetic activities. These results reveal that SynCAM 1 acts in developing neurons to shape migrating growth cones and contributes to the adhesive differentiation of their axo-dendritic contacts. |
|
Signaling by synaptogenic molecules. Biederer T, Stagi M.
AbstractMultiple signaling pathways initiate and specify the formation of synapses in the central nervous system. General principles that organize nascent synapses have emerged from the studies in multiple model organisms. These include the synapse-organizing roles of dedicated synaptic adhesion molecules, synaptic signaling following receptor-ligand interactions, and the regulation of synapse formation by secreted molecules. Intracellularly, a range of effectors subsequently regulates signaling steps and cytoskeletal changes. Together, a blueprint of synapse formation is emerging into which these distinct signaling steps will need to be integrated temporally and spatially. |
 |
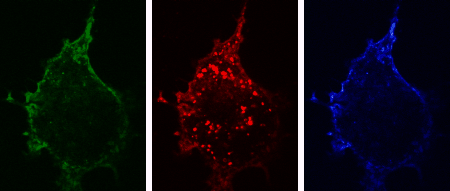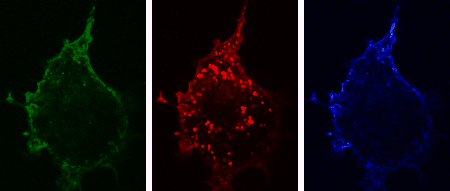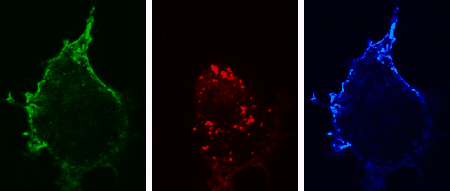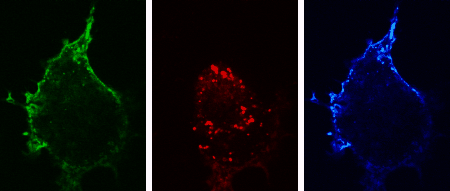
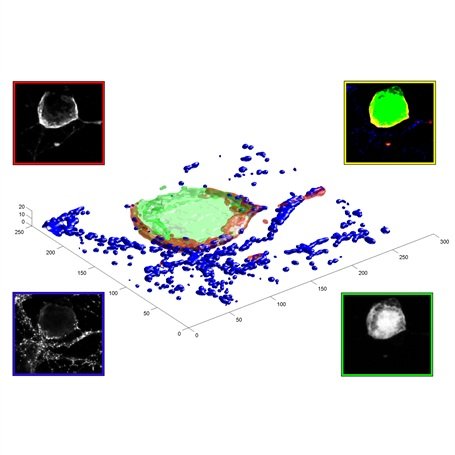 The three-dimensional reconstruction shows a HEK293 cells expressing SynCAM 2 on its surface (green),
with neuronal SynCAM 1 (red) bound and retained atop. Surface contact sites with neurites also contain clustered synapsin puncta (blue), consistent with synapse organization mediated by synaptic adhesion molecules of the SynCAM family. |
SynCAMs organize synapses through heterophilic adhesion. * = Equal contribution
Fogel AI, Stagi M*,Akins MR*, Krupp AJ*, Stein V, Biederer T.
AbstractSynapses are asymmetric cell junctions with precisely juxtaposed presynaptic and postsynaptic sides. Transsynaptic adhesion complexes are thought to organize developing synapses. The molecular composition of these complexes, however, remains incompletely understood, precluding us from understanding how adhesion across the synaptic cleft guides synapse development. Here, we define two immunoglobulin superfamily members, SynCAM 1 and 2, that are expressed in neurons in the developing brain and localize to excitatory and inhibitory synapses. They function as cell adhesion molecules and assemble with each other across the synaptic cleft into a specific, transsynaptic SynCAM 1/2 complex. Additionally, SynCAM 1 and 2 promote functional synapses as they increase the number of active presynaptic terminals and enhance excitatory neurotransmission. The interaction of SynCAM 1 and 2 is affected by glycosylation, indicating regulation of this adhesion complex by posttranslational modification. The SynCAM 1/2 complex is representative for the highly defined adhesive patterns of this protein family, the four members of which are expressed in neurons in divergent expression profiles. SynCAMs 1, 2, and 3 each can bind themselves, yet preferentially assemble into specific, heterophilic complexes as shown for the synaptic SynCAM 1/2 interaction and a second complex comprising SynCAM 3 and 4. Our results define SynCAM proteins as components of novel heterophilic transsynaptic adhesion complexes that set up asymmetric interactions, with SynCAM proteins contributing to synapse organization and function. |
|
TREM2-transduced myeloid precursors mediate nervous tissue debris clearance and facilitate recovery in an animal model of multiple sclerosis. Takahashi K, Prinz M, Stagi M., Chechneva O, Neumann H.
AbstractIn multiple sclerosis, inflammation can successfully be prevented, while promoting repair is still a major challenge. Microglial cells, the resident phagocytes of the central nervous system (CNS), are hematopoietic-derived myeloid cells and express the triggering receptor expressed on myeloid cells 2 (TREM2), an innate immune receptor. Myeloid cells are an accessible source for ex vivo gene therapy. We investigated whether myeloid precursor cells genetically modified to express TREM2 affect the disease course of experimental autoimmune encephalomyelitis (EAE), an animal model of multiple sclerosis. EAE was induced in mice by immunization with a myelin autoantigen. Intravenous application of TREM2-transduced bone marrow-derived myeloid precursor cells at the EAE peak led to an amelioration of clinical symptoms, reduction in axonal damage, and prevention of further demyelination. TREM2-transduced myeloid cells applied intravenously migrated into the inflammatory spinal cord lesions of EAE-diseased mice, showed increased lysosomal and phagocytic activity, cleared degenerated myelin, and created an anti-inflammatory cytokine milieu within the CNS. Intravenously applied bone marrow-derived and TREM2-tranduced myeloid precursor cells limit tissue destruction and facilitate repair within the murine CNS by clearance of cellular debris during EAE. TREM2 is a new attractive target for promotion of repair and resolution of inflammation in multiple sclerosis and other neuroinflammatory diseases. |
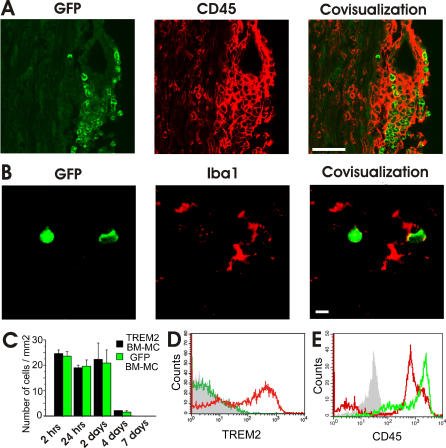 Migration of TREM2-Transduced Myeloid Cells into EAE Lesions
|
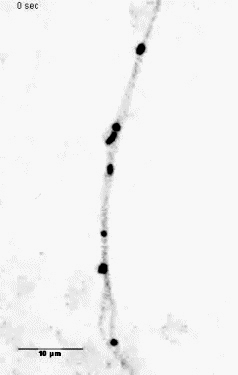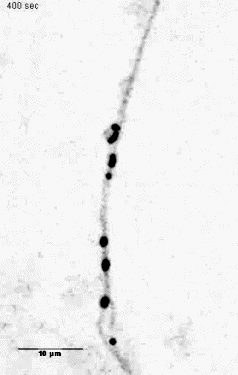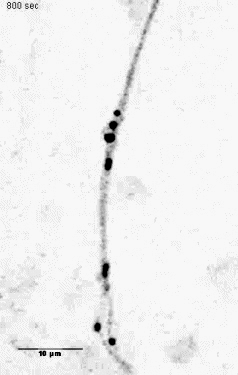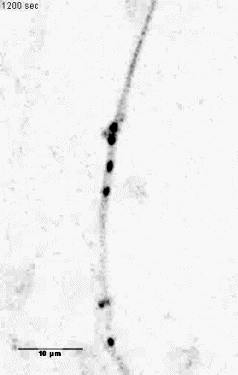 Mitochondria tracking of an axonal segment of a cultured hippocampal neuron
transfected with a mitochondrial targeting sequence tagged with YFP. |
Unloading kinesin transported cargoes from the tubulin track via the inflammatory c-Jun N-terminal kinase pathway. * = Equal contribution
Stagi M*, Gorlovoy P*, Larionov S, Takahashi K, Neumann H.
AbstractAxonal transport of mitochondria and synaptic vesicle precursors via kinesin motor proteins is essential to keep integrity of axons and synapses. Disturbance of axonal transport is an early sign of neuroinflammatory and neurodegenerative diseases. Treatment of cultured neurons by the inflammatory cytokine tumor necrosis factor-alpha (TNF) stimulated phosphorylation of c-Jun N-terminal kinase (JNK) in neurites. TNF treatment induced dissociation of the heavy chain kinesin family-5B (KIF5B) protein from tubulin in axons but not cell bodies as determined by lifetime-based F�rster resonance energy transfer (FRET) analysis. Dissociation of KIF5B from tubulin after TNF treatment was dependent on JNK activity. Furthermore, TNF inhibited axonal transport of mitochondria and synaptophysin by reducing the mobile fraction via JNK. Thus, TNF produced by activated glial cells in inflammatory or degenerative neurological diseases acts on neurites by acting on the kinesin-tubulin complex and inhibits axonal mitochondria and synaptophysin transport via JNK. |
|
LPS receptor (CD14): a receptor for phagocytosis of Alzheimer's amyloid peptide. * = Equal contribution
Liu Y, Walter S*, Stagi M.*, Cherny D, Letiembre M, Schulz-Schaeffer W, Heine H, Penke B, Neumann H, Fassbender K.
AbstractThe amyloid beta peptide 42 (Abeta(42)) plays a key role in neurotoxicity in Alzheimer's disease. Mononuclear phagocytes, i.e. microglia, have the potential to clear Abeta by phagocytosis. Recently, the lipopolysaccharide (LPS) receptor CD14 was shown to mediate phagocytosis of bacterial components and furthermore to contribute to neuroinflammation in Alzheimer's disease. Here, we investigated whether this key innate immunity receptor can interact with Abeta(42) and mediate phagocytosis of this peptide. Using flow cytometry, confocal microscopy and two-photon fluorescence lifetime imaging (FLIM) combined with fluorescence resonance energy transfer (FRET), we demonstrated a direct molecular interaction in the range of a few nanometers between Abeta(42) and CD14 in human CD14-transfected Chinese hamster ovary cells. Investigations using cells that were genetically deficient for this receptor showed that in 30 minutes exogenous Abeta(42) added to cultured primary microglial cells was phagocytosed into the cytoplasmic compartment in a CD14-dependent manner. This phagocytosis occurred at Abeta(42) concentration ranges that were considerably lower than the threshold to activate a cellular inflammatory reaction. In contrast, there was no association of CD14 to microglial internalization of microbeads. In complementary clinical experiments, we detected a pronounced CD14 immunoreactivity on parenchymal microglia spatially correlated to characteristic Alzheimer's disease lesion sites in brain sections of Alzheimer's disease patients but not in brain sections of control subjects. By showing a close interaction between CD14 and Abeta(42), demonstrating a direct role of CD14 in Abeta(42) phagocytosis, and detecting CD14-specific staining in brains of Alzheimer's disease patients, our results indicate a role of the LPS receptor in the pathophysiology of Alzheimer's disease, which could be of therapeutic relevance. |
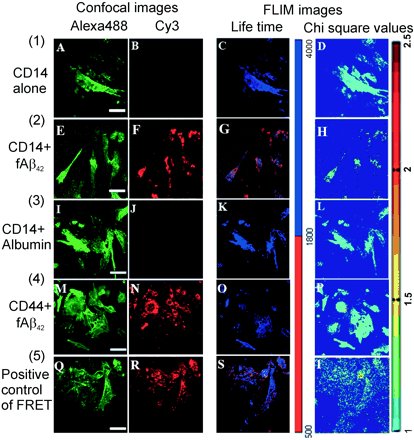 Analysis of fluorescence lifetime distribution.
FRET between Alexa488-stained CD14 and Cy3-stained biotinylated ABeta42 |
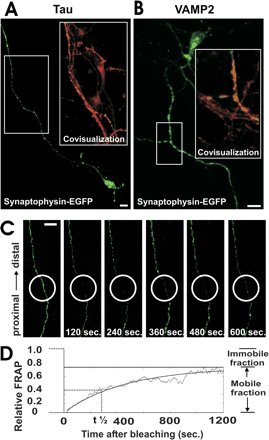 Axonal expression and FRAP analysis of synaptophysin-EGFP. a, Neuron transfected with synaptophysin-EGFP (green)
and immunolabeled with antibodies directed against tau (red). Synaptophysin-EGFP is localized in the axon. |
Breakdown of axonal synaptic vesicle precursor transport by microglial nitric oxide. Stagi M, Dittrich PS, Frank N, Iliev AI, Schwille P, Neumann H.
AbstractThe mechanism of axonal injury in inflammatory brain diseases is still unclear. Increased microglial production of nitric oxide (NO) is a common early sign in neuroinflammatory diseases. We found by fluorescence correlation spectroscopy that synaptophysin tagged with enhanced green fluorescence protein (synaptophysin-EGFP) moves anterogradely in axons of cultured neurons. Activated microglia focally inhibited the axonal movement of synaptophysin-EGFP in a NO synthase-dependent manner. Direct application of a NO donor to neurons resulted in inhibition of axonal transport of synaptophysin-EGFP and synaptotagmin I tagged with EGFP, mediated via phosphorylation of c-jun NH2-terminal kinase (JNK). Thus, overt production of reactive NO by activated microglia blocks the axonal transport of synaptic vesicle precursors via phosphorylation of JNK and could cause axonal and synaptic dysfunction. |